 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicholas Leventis | -- | 2684 | 2022-04-01 19:33:48 | | | |
| 2 | Amina Yu | -30 word(s) | 2654 | 2022-04-02 03:37:32 | | |
Video Upload Options
The term “aerogel” describes a certain class of low-density solid materials with a high open porosity. Aerogels can be considered to be a subclass of the much broader domain of porous materials; however, they are distinguished from, for example, blown foams, because they are prepared in a completely different manner—namely, by drying wet gels in a way that preserves nearly all of their volume in the final dry form.
1. The Chemistry of Polyurea Aerogels
1.1. The Chemistry of the Isocyanate Group
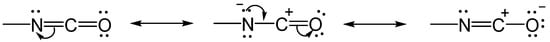
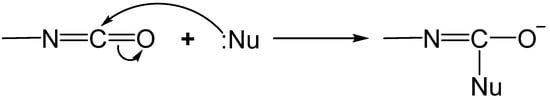

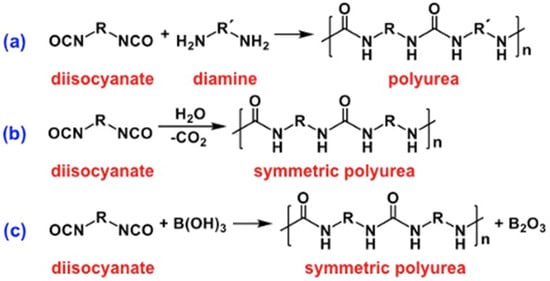
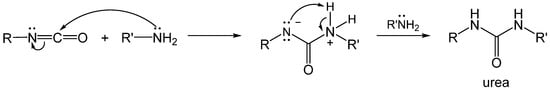
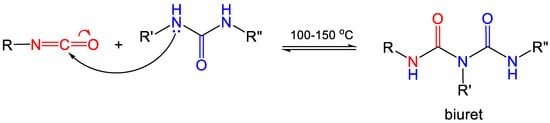
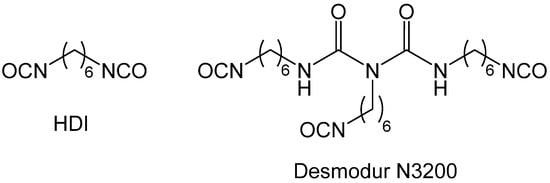
1.2. Formation of Polyurea Aerogels via Reaction of the –N=C=O with Amines
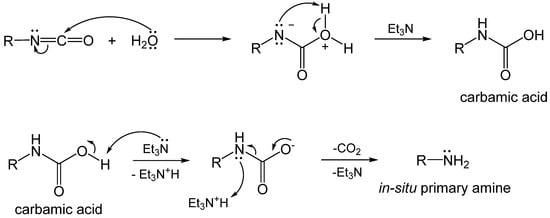
1.3. Formation of Polyurea Aerogels via Reaction of –N=C=O with Water
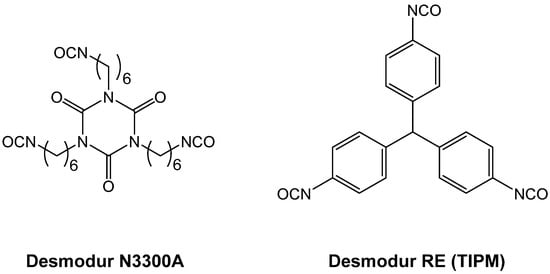
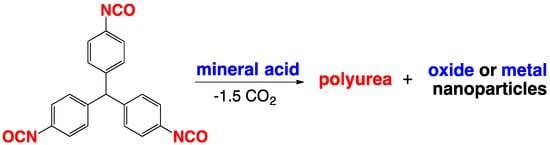
1.4. Formation of Polyurea Aerogels via Reaction of –N=C=O with Mineral Acids
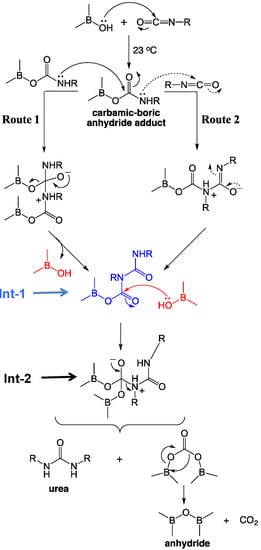
2. Translating the Polymerization Chemistry into Aerogels
2.1. The Gelation Process and Nanomorphology
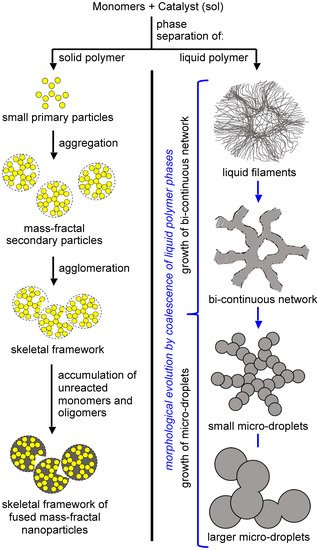
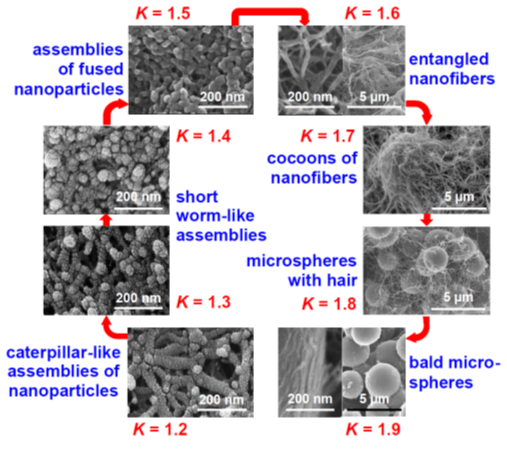
Figure 13. Eight (8) nanomorphology groups identified from 188 formulations of Desmodur N3300A/water-derived polyurea aerogels prepared using 8 different solvents (i.e., acetone, acetonitrile, nitromethane, propylene carbonate, THF, DMF, 2-butanone, and ethyl acetate). K-index is defined as the ratio of the water contact angle to the percentage of porosity for a given material. Each morphological group shown in this figure was associated with a unique K-index value [21].
These skeletal morphologies of Figure 13 translate directly into a wide range of surface areas, texture-induced hydrophobicity, and diverse mechanical properties that include superelasticity and shape-memory effects.
2.2. Molding, form Factors, and the Drying Process
References
- Saunders, J.H.; Frisch, K.C. Polyurethane Chemistry and Technology: Chemistry; Interscience Publishers: Hoboken, NJ, USA, 1964; pp. 63–118.
- Stevens, M.P. Polymer Chemistry. An Introduction; Oxford University Press: New York, NY, USA, 1990.
- Davis, T.L.; Ebersole, F. Relative velocities of reaction of amines with phenyl isocyanate. J. Am. Chem. Soc. 1934, 56, 885–886.
- Neumann, W.; Fischer, P. Carbodiimide aus Isocyanaten. Angew. Chem. 1962, 74, 801–806.
- Available online: https://coatings.specialchem.com/product/r-covestro-desmodur-n-3200 (accessed on 26 February 2022).
- Wicks, Z.W., Jr.; Jones, F.N.; Pappas, S.P.; Wicks, D.A. Organic Coatings, Science & Technology, 3rd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007.
- Odian, G. Principles of Polymerization; Wiley-Interscience: New York, NY, USA, 2004.
- Dodge, J. Polyurethanes and Polyureas. In Synthetic Methods in Step-Growth Polymers; Rogers, M.E., Long, T.E., Eds.; Wiley: New York, NY, USA, 2003; pp. 197–263.
- Leventis, N.; Sotiriou-Leventis, C.; Saeed, A.M.; Donthula, S.; Majedi Far, H.; Rewatkar, P.M.; Kaiser, H.; Robertson, J.D.; Lu, H.; Churu, G. Nanoporous polyurea from a triisocyanate and boric acid: A paradigm of a general reaction pathway for isocyanates and mineral acids. Chem. Mater. 2016, 28, 67–78.
- Aries, R.S. Polymers from Boric Acid and Organic Diisocyanates. U.S. Patent No 2,945,841, 19 July 1960.
- Xiao, H.; Xiao, H.X.; Frisch, K.C.; Malwitz, N. Kinetic studies of the reactions between isocyanates and carboxylic acids. High Perform. Polym. 1994, 6, 235–239.
- Sorenson, W.R. Reaction of an isocyanate and a carboxylic acid in dimethyl sulfoxide. J. Org. Chem. 1959, 24, 978–980.
- Trappe, V.; Prasad, V.; Cipelletti, L.; Segre, P.N.; Weitz, D.A. Jamming phase diagrams for attractive particles. Nature 2001, 411, 772–775.
- Lu, P.J.; Zaccarelli, E.; Ciulla, F.; Schofield, A.B.; Sciortino, F.; Weitz, D.A. Gelation of particles with short-range attraction. Nature 2008, 499, 453–503.
- Rouwhorst, J.; Ness, C.; Stoyanov, S.; Zaccone, A.; Schall, P. Nonequilibrium continuous phase transition in colloidal gelation with short-range attraction. Nature Commun. 2020, 11, 3558.
- Bang, A.; Buback, C.; Sotiriou-Leventis, C.; Leventis, N. Flexible aerogels from hyperbranched polyurethanes: Probing the role of molecular rigidity with poly(urethane acrylates) versus poly(urethane norbornenes). Chem. Mater. 2014, 26, 6979–6993.
- Papastergiou, M.; Kanellou, A.; Chriti, D.; Raptopoulos, G.; Paraskevopoulou, P. Poly(urethane-acrylate) aerogels via radical polymerization of dendritic urethane-acrylate monomers. Materials 2018, 11, 2249.
- Mohite, D.P.; Larimore, Z.J.; Lu, H.; Mang, J.T.; Sotiriou-Leventis, C.; Leventis, N. Monolithic hierarchical fractal assemblies of silica nanoparticles cross-linked with polynorbornene via ROMP: A structure-property correlation from molecular to bulk through nano. Chem. Mater. 2012, 24, 3434–3448.
- Mohite, D.P.; Mahadik-Khanolkar, S.; Luo, H.; Lu, H.; Sotiriou-Leventis, C.; Leventis, N. Polydicyclopentadiene aerogels grafted with PMMA: II. Nanoscopic characterization and origin of macroscopic deformation. Soft Matter 2013, 9, 1531–1539.
- Chidambareswarapattar, C.; McCarver, P.M.; Luo, H.; Lu, H.; Sotiriou-Leventis, C.; Leventis, N. Fractal multiscale nanoporous polyurethanes: Flexible to extremely rigid aerogels from multifunctional small molecules. Chem. Mater. 2013, 25, 3205–3224.
- NEW REFERENCE: Taghvaee, T.; Donthula, S.; Rewatkar, P.M.; Majedi Far, H.; Sotiriou-Leventis, C.; Leventis, N. K-index: Descriptor, predictor, and correlator of complex nanomorphology to other material properties. ACS Nano 2019, 13, 3677–3690.
- Malakooti, S.; ud Doulah, A.B.M.S.; Ren, Y.; Kulkarni, V.N.; Soni, R.U.; Edlabadkar, V.A.; Zhang, R.; Vivod, S.L.; Chariklia Sotiriou-Leventis, C.; Leventis, N.; et al. Meta-aerogels: Auxetic shape-memory polyurethane aerogels. ACS Appl. Polym. Mater. 2021, 3, 5727–5738.
- Członka, S.; Bertino, M.F.; Kośny, J.; Shukla, N. Freeze-drying method as a new approach to the synthesis of polyurea aerogels from isocyanate and water. J. Sol-Gel Sci. Technol. 2018, 87, 685–695.
- Steiner, S.A., III; Griffin, J.S.; Wunsch, B.H.; Schneider, J.N. Systems and Methods for Producing Aerogel Materials. U.S. Patent No 10,563,035 B2, 18 February 2020.

