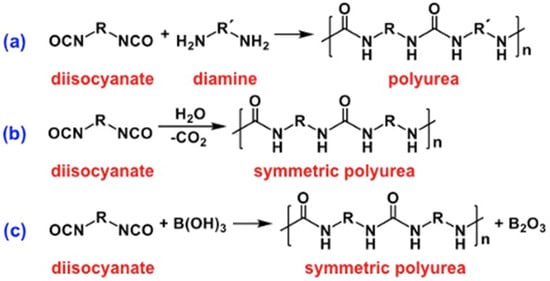
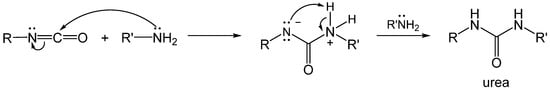
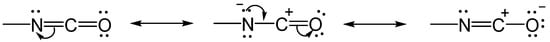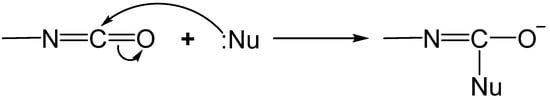2.1. The Gelation Process and Nanomorphology
The polymerization processes described in
Section 1 turn soluble monomers into insoluble polymers that comprise the skeletal framework of the aerogel. The morphology of the skeletal framework of polyurea aerogels can be very diverse, even within the well-defined chemical composition of a specific polymer. This morphological diversity arises from parameters such as the molecular structure of the monomers (e.g., aromatic versus aliphatic, rigid versus flexible, difunctional versus polyfunctional, the functional group density at the monomer molecular level), the solubility properties of the medium (e.g., polarity, ability to develop dispersion forces, and hydrogen bonding), the concentrations of the monomers and the catalyst, and the gelation temperature.
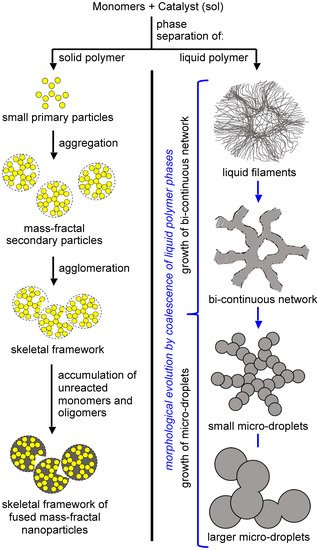
The formation of an insoluble polymer means phase separation, which can be of either solid primary particles (
Figure 12, left branch), or of an oily phase of insoluble oligomers (
Figure 12, right branch) [
34,
35,
36]. In the first case, the size of the primary particles decreases as the functionality and functional-group density of the monomers increases, leading quickly to a highly crosslinked, insoluble polymer. This has been demonstrated well with poly(urethane acrylate) aerogels derived from trifunctional versus nine-functional (nonafunctional) star and dendritic monomers, respectively; the two types of monomers, made using the same rigid aromatic triisocyanate core, yielded primary particles with radii equal to 17 nm versus 7 nm, respectively [
37,
38].
Figure 12. Phase separation and growth processes involved in the formation of the skeletal network of gels.
Phase-separated primary particles move randomly in the sol, and upon collision with one another they may become chemically connected via reaction with yet-unreacted surface functional groups inherited from the monomers. If the reaction is fast (and, thus, the sticking probability for each collision is high), the kinetics of network growth are limited only by diffusion, and the process is referred to as diffusion-limited aggregation. Such aggregates are mass fractal objects [
36], and their size is limited by the fact that their density falls rapidly with distance from the nucleation point at their center; this is to say that beyond a certain size there is not enough mass in their outer “layers” to support further growth. These fractal aggregates of primary particles are referred to as secondary particles. On geometric grounds, the pore sizes within secondary particles are in the same order as the primary particles, and typically they fall in the mesopore range (2–50 nm). Secondary particles also randomly walk, albeit more slowly, eventually being connected to one another via chemical bonding to form higher aggregates. The liquid sol turns into a solid gel once secondary particles, or higher aggregates, have formed a continuous interconnected three-dimensional path spanning the entire volume of the liquid within a mold. At that point, assuming that all monomers have been consumed, the skeletal network consists of interconnected secondary particles or higher aggregates. Experimentally, primary particles are identified, and their size is estimated using small-angle X-ray or neutron-scattering data (SAXS or SANS, respectively); alternatively, the primary particle size is calculated from skeletal density and N
2 sorption data via
r = 3/(
ρs ×
σ), where
r is the particle radius,
ρs is the skeletal density from He pycnometry; and
σ is the BET surface area from N
2 sorption porosimetry. The primary particle size obtained via the two methods should be roughly equal. This situation is very common with oxide aerogels, and is also encountered with several polyurea aerogels. If unreacted monomers or soluble oligomers remain in the pores after gelation, these species eventually find their way onto the protoskeletal framework, where they can accumulate via reaction with still-live functional groups on the surface of the framework. Eventually, accumulation of monomers and/or oligomers fills the void space within the secondary particles and, experimentally, the particle size calculated from skeletal density and N
2 sorption data reflects a size closer to that of the secondary particles as determined via SAXS or SANS. Under SEM, these structures appear as if they consist of random assemblies of particles, with a layer of polymer cast over the entire network [
37]. The topography of such an arrangement actually resembles that of polymer-crosslinked oxide aerogels, in which silica primary particles are embedded within a crosslinker-derived polymer that fills the space within the aerogel’s secondary particles [
39,
40]. Comparisons of particle sizes determined from skeletal density/N
2 sorption data and SAXS have proven useful in understanding the growth mechanisms underlying the formation of polymeric aerogels in general.
An interesting variation of the theme described in the previous paragraph is encountered when, within a series of aerogel samples of variable density, the particle size determined from skeletal density/N
2 sorption data matches the primary particle size from SAXS for high-density aerogels, but diverges—sometimes by orders of magnitude—at lower densities. This type of density-dependent trend is attributed to a sol-concentration-dependent gelation mechanism, and is typically accompanied by obvious morphological changes in SEM and large changes in the BET surface area [
41].
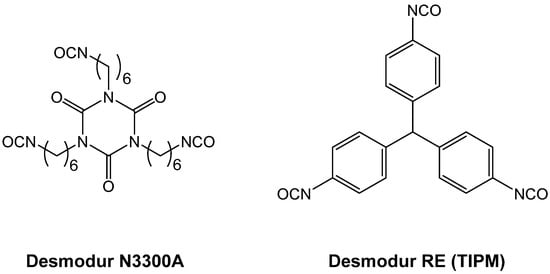
Phase separation of oligomers as an oily liquid (Figure 12, right branch) occurs with large flexible monomers with low functional-group density. The “oil” continues to react internally until it is solidified (arrested). Depending on the speed of solidification and the properties of the medium, the structural diversity that can be generated via this process can be extremely broad. For example, the oily phase may start out as thin, hair-like filaments that evolve into small droplets embedded within fibers; among other possibilities, the latter morphology may evolve into large particles with or without fibers emanating from their surface. By choosing the right monomers and adjusting the reaction rate (e.g., via adjustment of monomer concentration, or by using an appropriate catalyst), any skeletal framework with these morphologies can be “frozen” (kinetically trapped) in place before it has time to evolve into the next step. Polyurea aerogels are an extremely versatile class of materials that enable observation and, in some cases, design of those morphologies in the final products. For example, Figure 13 shows a very large array of nanostructures, all with identical chemical composition, prepared via reaction of Desmodur N3300A (see Figure 9) and water (see Figure 4b) in various solvents at different monomer concentrations.
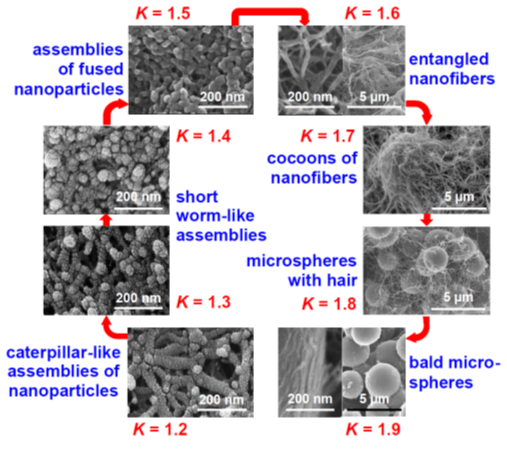
Figure 13. Eight (8) nanomorphology groups identified from 188 formulations of Desmodur N3300A/water-derived polyurea aerogels prepared using 8 different solvents (i.e., acetone, acetonitrile, nitromethane, propylene carbonate, THF, DMF, 2-butanone, and ethyl acetate). K-index is defined as the ratio of the water contact angle to the percentage of porosity for a given material. Each morphological group shown in this figure was associated with a unique K-index value [NEW REFERENCE: Taghvaee, T.; Donthula, S.; Rewatkar, P.M.; Majedi Far, H.; Sotiriou-Leventis, C.; Leventis, N. K-index: Descriptor, predictor, and correlator of complex nanomorphology to other material properties. ACS Nano 2019, 13, 3677–3690.]
These skeletal morphologies of Figure 13 translate directly into a wide range of surface areas, texture-induced hydrophobicity, and diverse mechanical properties that include superelasticity and shape-memory effects.