Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ivan Okhrimenko | -- | 1343 | 2022-04-01 15:58:21 | | | |
| 2 | Vicky Zhou | Meta information modification | 1343 | 2022-04-06 10:52:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Okhrimenko, I.; Borshchevskiy, V.; Bogorodskiy, A.; Burkatovskii, D.; , .; Gordeliy, V.; Gensch, T.; Voos, W.; Altschmied, J.; Haendeler, J.(. Mitochondrial Protein Import. Encyclopedia. Available online: https://encyclopedia.pub/entry/21291 (accessed on 24 July 2026).
Okhrimenko I, Borshchevskiy V, Bogorodskiy A, Burkatovskii D, , Gordeliy V, et al. Mitochondrial Protein Import. Encyclopedia. Available at: https://encyclopedia.pub/entry/21291. Accessed July 24, 2026.
Okhrimenko, Ivan, Valentin Borshchevskiy, Andrey Bogorodskiy, Dmitrii Burkatovskii, , Valentin Gordeliy, Thomas Gensch, Wolfgang Voos, Joachim Altschmied, Jojo (Judith) Haendeler. "Mitochondrial Protein Import" Encyclopedia, https://encyclopedia.pub/entry/21291 (accessed July 24, 2026).
Okhrimenko, I., Borshchevskiy, V., Bogorodskiy, A., Burkatovskii, D., , ., Gordeliy, V., Gensch, T., Voos, W., Altschmied, J., & Haendeler, J.(. (2022, April 01). Mitochondrial Protein Import. In Encyclopedia. https://encyclopedia.pub/entry/21291
Okhrimenko, Ivan, et al. "Mitochondrial Protein Import." Encyclopedia. Web. 01 April, 2022.
Copy Citation
Mitochondria play a critical role in providing energy, maintaining cellular metabolism, and regulating cell survival and death. To carry out these crucial functions, mitochondria employ more than 1500 proteins, distributed between two membranes and two aqueous compartments. An extensive network of dedicated proteins is engaged in importing and sorting these nuclear-encoded proteins into their designated mitochondrial compartments. Defects in this fundamental system are related to a variety of pathologies, particularly engaging the most energy-demanding tissues.
mitochondria
mitochondrial protein import
1. Introduction
In general, mitochondria are known as the powerhouses of cells since their major function is to produce ATP as an energy source. Besides that, mitochondria are required for the regulation of calcium homeostasis, lipid and amino acid metabolism, and biosynthesis of heme and iron-sulfur complexes. Over the last years, it has become evident that mitochondria are signaling organelles, which regulate processes like apoptotic cell death as well as nuclear transcription [1].
Mitochondrial functions decline with the aging process of the organism, as mitochondria are subjected to a variety of biochemical stress conditions. Those lead to the accumulation of damaged molecules that directly or indirectly interfere with the regular biochemical processes occurring in mitochondria. Most prominent in this background are oxidative stress reactions caused by the accumulation of reactive oxygen species (ROS). ROS are generated as byproducts during the metabolic reactions of the OXPHOS process, in addition to detrimental environmental impacts [2]. Mitochondrial biogenesis capacity declines at older age, in particular due to the accumulation of mutations in the mitochondrial and nuclear genome. These mutations lead to the generation of defective or aberrant enzymatic components of the mitochondrial structure and metabolism, resulting in mitochondrial and eventually cellular dysfunction. This general problem is exacerbated by a relatively error-prone mitochondrial genome replication and inefficient repair processes [3].
As cellular survival depends on the maintenance of protein function, also called proteostasis, an efficient protein biogenesis process is a prerequisite to providing the required proteins for all cellular functions. Due to the dual genetic origin of mitochondrial proteins, a portion is encoded in the mitochondria, but most are nuclear-encoded; efficient protein biosynthesis and subsequent import into the organelle are essential for mitochondrial function. In addition to the transport processes into the organelle, mitochondrial polypeptides—irrespective of their source—need to undergo proper folding and assembly into active enzymes or enzyme complexes. Thus, the biogenesis of mitochondrial proteins represents a very complex process that is also prone to errors and defects correlating with diseases. Of note, not only the proteins in the inner (IMM) and outer mitochondrial membranes (OMM) are required for proper complex assembly, but also the phospholipid composition of the mitochondrial membranes plays a critical role. For example, it has been demonstrated that the lipid composition changes with age; in particular, the content of the mitochondria-specific lipid cardiolipin declines, which may play a central role in age-related diseases [4]. Age-related accumulation of cholesterol in mitochondria is a proposed trigger of Alzheimer’s disease (AD) [5][6].
Stress- or age-related damage of individual proteins or even the full organelle is counteracted by an array of different protective processes. On the protein side, a variety of chaperone and protease enzymes help to refold or remove damaged polypeptides before they accumulate and induce malfunction. To deal with unfolded polypeptides, mitochondria contain the full set of Hsp70- and Hsp60-type chaperones, similar to their bacterial ancestors [7]. In addition, each mitochondrial subcompartment, including the membranes, contains protease enzymes that specifically digest polypeptides that could not be refolded or assembled into their respective enzyme complexes [8]. Failure of this protein quality control (PQC) system contributes to many human diseases [9]. Specific signaling processes from mitochondria to the nucleus, summarized under the expression “unfolded protein response (mtUPR)”, increase the mitochondrial PQC capacity by enhancing the expression of the respective chaperones and proteases. If these protective processes fail, defective mitochondria themselves seem to be removed by a regulated and specific form of autophagy, named mitophagy [10].
As mitochondrial functions and quality control are largely dependent on the import of proteins produced in the cytoplasm, disruptions in import create cascading effects in mitochondria leading to diminished metabolic function, increased production of ROS, and failures in regulated cell death response [11]. Mitochondrial dysfunction is increased particularly with older age. For several diseases, most importantly affecting organs that particularly depend on mitochondria, namely the brain and the heart, mitochondrial dysfunction has been broadly described [1][12][13][14][15].
Not surprisingly, a number of recent reviews describe different aspects of mitochondrial function and structure, in particular mitochondrial machineries for protein import and assembly [16]. Considerably fewer reviews can be found on the connection between mitochondrial protein import and aging-related diseases [17].
2. Mitochondrial Protein Import Machinery
Nuclear-encoded mitochondrial proteins typically contain signaling sequences that are recognized in the cytosol by the mitochondrial protein import machinery and transported into mitochondria in an ATP-dependent manner (for review, see [16]). Mitochondrial targeting sequences can either be N-terminal (e.g., subunit 8 of cytochrome c oxidase), internal (e.g., ADP/ATP carrier), or C-terminal (e.g., VDAC) [18][19][20][21]). N-terminal presequences are the most frequent and can be recognized by mitochondria import machinery either during or after translation [22]. After import, around 70% of presequences are cleaved by mitochondrial processing peptidases (MPP) [23]. Internal and C-terminal mitochondrial targeting sequences can only be recognized after protein translation is complete. Mitochondrial proteins employing internal or C-terminal targeting sequences are frequently hydrophobic and require assistance from chaperones such as Hsp70/90 to prevent misfolding and aggregation in the cytoplasm.
Mitochondrial protein recognition occurs at the translocase of the outer membrane (TOM) complex. The TOM component TOMM20 acts as a direct receptor for N-terminal signal sequences, while TOMM70 seems to be dedicated to the recognition of hydrophobic proteins with internal signal sequences. These hydrophobic proteins reach the mitochondrial surface, often bound by cytosolic chaperones that also can interact with TOMM70 [16]. After passing through pore formed by the TOM complex, proteins are sorted between TIM22, TIM23, SAM, or MIA machinery, depending on the protein destination and fold (Figure 1). Insertase OXA1L inserts mitochondria encoded proteins into IMM. In yeast, mitochondrial insertase Oxa1p additionally assists in inserting multi α-helical nuclear-encoded proteins, but the same behavior for human OXA1L was not yet verified [24][25].
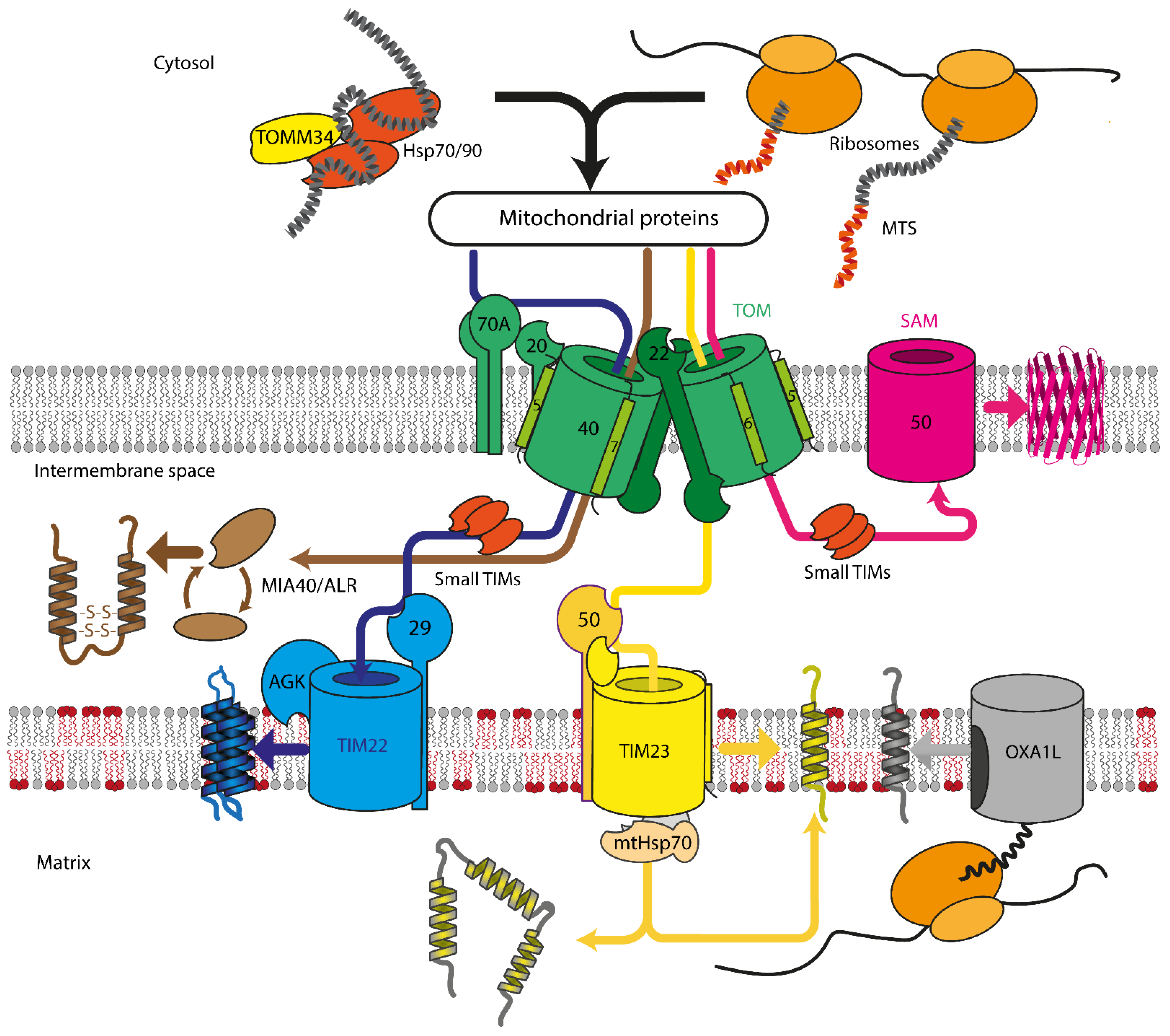
Figure 1. Overview of mitochondrial protein import in mammalian cells. Proteins are recognized by TOMM70A/TOMM20/TOMM22 and are imported either co- or post-translationally through the TOM complex, containing TOMM40, TOMM22, TOMM5, TOMM6, and TOMM7. Hydrophobic proteins employ Hsp70/90 complex with participation of TOMM34 to prevent misfolding in the cytoplasm. Inside the intermembrane space, depending on the nature and destination of the precursor protein, proteins are delivered to different compartments. β-barrels of the outer membrane are inserted into the outer mitochondrial membrane by SAM complex. Intermembrane space proteins with cysteine motifs are oxidized to the mature form by the MIA40/ALR system. Metabolite carriers are inserted into IMM by TIM22 complex, composed of TIMM22, TIMM29, and acylglycerol kinase (AGK). Other IMM and matrix proteins are inserted/transported by TIM23 complex. Primary TIMM23 pore is associated with TIMM50 (recognizes signals and interacts with TOM complex), TIMM44 with associated mtHsp70 (forming presequence-associated motor helping matrix protein transfer), or TIMM21 for protein release into IMM. Mitochondrial-encoded proteins are inserted into the IMM by OXA1L insertase from the matrix side.
In addition to signaling sequences, mitochondrial protein import efficiency is further enhanced by localization of mRNA encoding mitochondrial proteins near the OMM and its translation in the vicinity of the translocation machinery [26][27][28], thus bypassing the need for intermediate chaperone-mediated transport. The observed mRNA localization near OMM is guided either by the interaction of protein nascent chains with TOM complex or by the interaction between 3′ mRNA end, specific RNA-binding proteins (RBPs), and OMM proteins. In human cells, RBPs are only incompletely described, but some proteins were found to co-localize with mRNA on the OMM surface, e.g., CluH and PUM [29][30]. Recent advances in proximity labeling identified several new RBPs [31] on the OMM surface, including SYNJ2BP, which persists on the OMM surface after disruption of protein synthesis and binds several mRNAs of OXPHOS proteins. RBPs’ participation in mitochondrial protein transport presents another potential avenue of protein import disruption caused by mutations/aging/environmental factors.
References
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284.
- Harman, D. The Free Radical Theory of Aging. Antioxid. Redox Signal. 2003, 5, 557–561.
- Park, C.B.; Larsson, N.-G. Mitochondrial DNA Mutations in Disease and Aging. J. Cell Biol. 2011, 193, 809–818.
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-Dependent Decline in the Cytochrome c Oxidase Activity in Rat Heart Mitochondria: Role of Cardiolipin. FEBS Lett. 1997, 406, 136–138.
- Garcia-Euiz, C.; Mari, M.; Coiell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañes, G.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Health and Disease. Histol. Histopathol. 2009, 24, 117–132.
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s Disease and Their Potential Role in Alzheimer’s Proteostasis. Exp. Neurol. 2020, 330, 113–321.
- Voos, W. Chaperone–Protease Networks in Mitochondrial Protein Homeostasis. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 388–399.
- Quirós, P.M.; Langer, T.; López-Otín, C. New Roles for Mitochondrial Proteases in Health, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359.
- Cenini, G.; Voos, W. Role of Mitochondrial Protein Quality Control in Oxidative Stress-Induced Neurodegenerative Diseases. Curr. Alzheimer Res. 2016, 13, 164–173.
- Rüb, C.; Wilkening, A.; Voos, W. Mitochondrial Quality Control by the Pink1/Parkin System. Cell Tissue Res. 2017, 367, 111–123.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Green, D.R.; Van Houten, B. SnapShot: Mitochondrial Quality Control. Cell 2011, 147, 950–950.e1.
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407.
- Simon, D.K.; Johns, D.R. Mitochondrial Disorders: Clinical and Genetic Features. Annu. Rev. Med. 1999, 50, 111–127.
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152.
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714.
- Palmer, C.S.; Anderson, A.J.; Stojanovski, D. Mitochondrial Protein Import Dysfunction: Mitochondrial Disease, Neurodegenerative Disease and Cancer. FEBS Lett. 2021, 595, 1107–1131.
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644.
- Van Kuilenburg, A.B.P.; Muijsers, A.O.; Demol, H.; Dekker, H.L.; Van Beeumen, J.J. Human Heart Cytochrome c Oxidase Subunit VIII Purification and Determination of the Complete Amino Acid Sequence. FEBS Lett. 1988, 240, 127–132.
- Pfanner, N.; Hoeben, P.; Tropschug, M.; Neupert, W. The Carboxyl-Terminal Two-Thirds of the ADP/ATP Carrier Polypeptide Contains Sufficient Information to Direct Translocation into Mitochondria. J. Biol. Chem. 1987, 262, 14851–14854.
- Jores, T.; Klinger, A.; Groß, L.E.; Kawano, S.; Flinner, N.; Duchardt-Ferner, E.; Wöhnert, J.; Kalbacher, H.; Endo, T.; Schleiff, E.; et al. Characterization of the Targeting Signal in Mitochondrial β-Barrel Proteins. Nat. Commun. 2016, 7, 12036.
- Becker, T.; Song, J.; Pfanner, N. Versatility of Preprotein Transfer from the Cytosol to Mitochondria. Trends Cell Biol. 2019, 29, 534–548.
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.P.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global Analysis of the Mitochondrial N-Proteome Identifies a Processing Peptidase Critical for Protein Stability. Cell 2009, 139, 428–439.
- Hell, K.; Neupert, W.; Stuart, R.A. Oxa1p Acts as a General Membrane Insertion Machinery for Proteins Encoded by Mitochondrial DNA. EMBO J. 2001, 20, 1281–1288.
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA 1L Mutations Cause Mitochondrial Encephalopathy and a Combined Oxidative Phosphorylation Defect. EMBO Mol. Med. 2018, 10, e9060.
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26.
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346, 748–751.
- Bogorodskiy, A.; Okhrimenko, I.; Maslov, I.; Maliar, N.; Burkatovskii, D.; von Ameln, F.; Schulga, A.; Jakobs, P.; Altschmied, J.; Haendeler, J.; et al. Accessing Mitochondrial Protein Import in Living Cells by Protein Microinjection. Front. Cell Dev. Biol. 2021, 9, 1794.
- Gao, J.; Schatton, D.; Martinelli, P.; Hansen, H.; Pla-Martin, D.; Barth, E.; Becker, C.; Altmueller, J.; Frommolt, P.; Sardiello, M.; et al. CLUH Regulates Mitochondrial Biogenesis by Binding MRNAs of Nuclear-Encoded Mitochondrial Proteins. J. Cell Biol. 2014, 207, 213–223.
- Ravanidis, S.; Doxakis, E. RNA-Binding Proteins Implicated in Mitochondrial Damage and Mitophagy. Front. Cell Dev. Biol. 2020, 8, 372.
- Qin, W.; Myers, S.A.; Carey, D.K.; Carr, S.A.; Ting, A.Y. Spatiotemporally-Resolved Mapping of RNA Binding Proteins via Functional Proximity Labeling Reveals a Mitochondrial MRNA Anchor Promoting Stress Recovery. Nat. Commun. 2021, 12, 4980.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
06 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No