Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nandini Dey | -- | 4131 | 2022-03-31 11:52:53 | | | |
| 2 | Rita Xu | + 1 word(s) | 4132 | 2022-03-31 12:05:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dey, N.; De, P.; Aske, J.; , . Bête Noire of Chemotherapy and Targeted Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/21216 (accessed on 29 June 2026).
Dey N, De P, Aske J, . Bête Noire of Chemotherapy and Targeted Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/21216. Accessed June 29, 2026.
Dey, Nandini, Pradip De, Jennifer Aske, . "Bête Noire of Chemotherapy and Targeted Therapy" Encyclopedia, https://encyclopedia.pub/entry/21216 (accessed June 29, 2026).
Dey, N., De, P., Aske, J., & , . (2022, March 31). Bête Noire of Chemotherapy and Targeted Therapy. In Encyclopedia. https://encyclopedia.pub/entry/21216
Dey, Nandini, et al. "Bête Noire of Chemotherapy and Targeted Therapy." Encyclopedia. Web. 31 March, 2022.
Copy Citation
Tumor cells struggle to survive following treatment. The struggle ends in either of two ways. The drug combination used for the treatment blocks the proliferation of tumor cells and initiates apoptosis of cells, which is a win for the patient, or tumor cells resist the effect of the drug combination used for the treatment and continue to evade the effect of anti-tumor drugs, which is a bête noire of therapy. Cancer-associated fibroblasts are the most abundant non-transformed element of the microenvironment in solid tumors. Tumor cells play a direct role in establishing the cancer-associated fibroblasts’ population in its microenvironment.
cancer-associated fibroblasts
resistance
chemotherapy
1. Introduction
Cancer-associated fibroblasts (CAFs) within the tumor microenvironment (TME) are non-transformed, tumor-cell-activated heterogeneous populations of cells having multiple origins and functions [1][2]. Detailed descriptions of the origin, functions, interactions with tumor cells, and heterogeneity of CAFs were previously provided by the researchers elsewhere [3][4]. CAFs are activated by tumor cells in their favor. Once activated in an established tumor, CAFs act as crucial supporters of tumor growth, progression, and response to treatment.
The functions of CAFs in an established tumor include the following: (1) ECM (extracellular matrix) remodeling via collagenolysis to promote invasion and EMT (endothelial–mesenchymal transition); (2) increasing tissue stiffness to initiate angiogenic resistance and immune suppression; (3) induction of tumor angiogenesis; (4) secretomic induction of EMT by TGFbeta; (5) increasing secretomic factors of tumor-promoting or immune-suppressing ligands such as hepatocyte growth factor; fibroblast growth factors 1 and 2; stromal cell-derived factor 1 (SDF1/CXCL12); chemokine (C-C motif) ligands (CCL) 2, 5, 7, and 16; interleukin 6/8; and platelet derived growth factor; (6) metabolic reversal of reverse Warburg effect (non-glycolysis in tumor cells, glycolysis in stroma cells) and ‘lactate shuttle’ effect; (7) immune evasion via activation of M2 macrophages (CD163 positive); (8) inhibition of apoptosis in tumor cells; (9) activation of many of pro-proliferative tumor cell signaling; (10) immune reprogramming and antigen presentation; (11) adaptation to oxidative stress and hypoxic response; (12) promotion of stemness-promoting signals; (13) promotion of metastasis-associated phenotypes; (14) attenuation of drug response [1][5][6][7][8][9][10][11][12][13][14][15][16][17][18].
The range of functions of CAFs is comprehensive, and the actions of CAFs are contextual. The interactions of CAFs with tumor cells and TME components change with the evolution of the tumor, its metastatic progression, and its response to therapy. In summary, the functions of CAFs are structured to assist and promote tumor cells via direct and indirect interactions. Thus, CAFs form a centralized communication network within the TME that favors tumor cell growth, metastasis, and resistance to drug treatment [19]. The versatility of the functions of CAFs’ make them abettors of drug resistance and identifies them as prospective anti-tumor therapy targets [20][21].
2. CAF Heterogeneity and Resistance to Chemotherapy in Solid Tumors
2.1. CAF Heterogeneity
CAFs are heterogeneous in terms of their origin in different organ-type cancers, as well as in the progression of the disease. The heterogeneous subpopulations of CAFs, such as myoblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs), have been extensively studied in fibroinflammatory PDAC disease characterized by dense and highly proliferating desmoplastic stroma. In fact, Li et al. identified genes associated with the differentiation of myCAFs and iCAFs [22][23][24]. Adipose-derived MSCs (AD-MSCs) have been shown to possess a high multilineage potential and self-renewal capacity and were reported as the CAF sources in PDAC by Miyazaki et al. [24]. Their study identified that AD-MSCs could differentiate into distinct CAF subtypes, myCAFs and iCAFs, depending on the different co-culture conditions in vitro. The diverse functions of iCAFs and myCAFs have also been reported in cholangiocarcinoma; breast cancers; prostate, head, and neck squamous cell carcinoma; and bladder and colon cancers. The diversity of CAF subpopulations was also recently reported to promote the growth of cholangiocarcinoma, wherein hepatic stellate cells (HSC) are the primary cause of CAF differentiation into myCAFs and iCAFs [25]. The hyaluronan synthase 2 myCAFs, but not type I collagen-expressing myCAFs, promoted tumor progression, while HGF-expressing iCAFs enhanced tumor growth via tumor-expressed MET, thereby directly linking CAFs to tumor cells. Another subset of CAFs, FAP+CAFs, were identified by Kieffer et al. in breast cancers that mediated immunosuppression and immunotherapy resistance via a positive feedback loop between specific CAF-S1 clusters and Tregs [26]. In prostate cancer, a differential mode of activation of iCAFs and myCAFs has been reported [27]. IL-1a/ELF3/YAP pathways are involved in iCAF differentiation, while TGF-beta1 induces myCAFs. One of the ways CAFs classically interact with the tumor cell EMT function was reported by Goulet et al. in bladder cancer, where IL-6 cytokine was found to be highly expressed in iCAFs, and its receptor IL-6R was found on RT4 bladder cancer cells [28]. Perhaps the most intriguing functional heterogeneity of CAFs was reported by Pan et al. in PDAC-CAF-exhibited organ-specific metastatic potential leading to different levels of heterogeneity of CAFs in different metastatic niches [29]. Several cell signaling pathways have been reported to be involved in the functioning of iCAFs and myCAFs, including the Hedgehog pathway [30]; Wnt pathway [31]; integrin a11B1 signaling [32]; cMET-HGF pathway [25]; IL-6 signaling [28]; EMT signaling via transcription factors SNAIL1, TWIST1, and ZEB1 [28]; and IL1B-mediated crosstalk [33]. Recently, Steele et al. reported that the Hedgehog pathway acts in a paracrine manner in PDAC, with ligands secreted by tumor cells signaling to stromal CAFs. The Hedgehog pathway activation is higher in PDPN+ alphaSMA+ myCAFs compared with iCAFs, and its inhibition impairs tumor growth by altering the fibroblast compartment in PDAC. Hedgehog pathway inhibition resulted in a reduction in myCAF numbers and a significant expansion of iCAFs, leading to an increase in the iCAF/myCAF ratio. As iCAFs are a source of inflammatory signals, the authors observed an increase in iCAFs upon Hedgehog inhibition, which correlated with changes in immune infiltration (significantly decreased CD8+ T cells and increased CD4+ T cells and CD25+CD4+ T cells; abundant FOXP3+ regulatory T cells) that are consistent with a more immunosuppressive pancreatic cancer microenvironment. The paracrine activation differentially elevated myCAFs compared with iCAFs, leading to favorable alterations of cytotoxic T cells and Tregs, causing increased immunosuppression [30]. Wnt signaling in CAFs represents a non-cell-autonomous mechanism for colon cancer progression [31]. Mose et al. reported Sfrp1 epithelial–mesenchymal transition phenotype induction in tumor cells without affecting tumor-intrinsic Wnt signaling, suggesting involvement of non-immune stromal cells. Low levels of Wnt signals induced the iCAF subtype, which in co-culture with organoids induced EMT, whereas high levels induced contractile myCAFs to attenuate the EMT phenotype.
The tumors with (1) an accumulation of stromal CAFs, (2) the presence of fibrotic stroma, (3) a high expression level of stroma signature genes, or (4) a high tumor/stroma ratio in the primary tumor are associated with poor prognosis in various cancers, including colon, gastric, esophagus, breast, NSCLC (non-small cell lung cancer), and liver cancers [34][35][36][37][38][39][40]. It is understood that chemotherapy’s limited effect (benefit) and the progression or recurrence of disease through therapy in many solid tumors are attributed to the development of resistance within tumor cells in support of the stroma. As a dominant component of tumor stroma, CAFs interact with both a tumor cell and the TME. The versatility of CAF functions and their several modes of interaction with tumor cells and all components of stroma (ECM and cells of the TME) indicate that a metastasis or progression of disease following treatment is aided and abetted by CAFs. Once a therapy-resistive circuitry is established between tumor cells and the CAFs of the stroma, tumor-centric therapy alone essentially becomes insufficient. Figure 1 presents the distribution pattern of the types of resistance to chemotherapy based on specific mediators of CAF functions in solid tumors. The four types of mediators of action employed by CAFs to orchestrate the development of resistance to chemotherapy are presented in the cartoon. The most common mode of interaction is a paracrine, wherein CAFs signal to either tumor cells or other components of the TME via characteristic secretomes. In addition to the involvement of characteristic secretomes, exosomal cargos delivering different miRNAs that target various cell signaling proteins are common mediators of CAFs. The paracrine mode of action of CAFs is the predominant form of action, represented by six types of organ tumors (organ tumors are indicated by their respective ribbon colors, as presented in the figure legends). CAF crosstalk with tumor cells, and the TME occurs via exosomal cargo, imparting resistance to four organ cancers. The extracellular vesicle, secretome, and autocrine or paracrine modes are much less involved in the modes of action (Figure 1). The sizes of the boxes indicate the number of studies in each box. Among resistance to different types of chemotherapies, cisplatin resistance has been found to be very common, which is involved in both paracrine and exosomal cargo modes of action (the shapes in the inset indicate the types of resistances in different tumors).
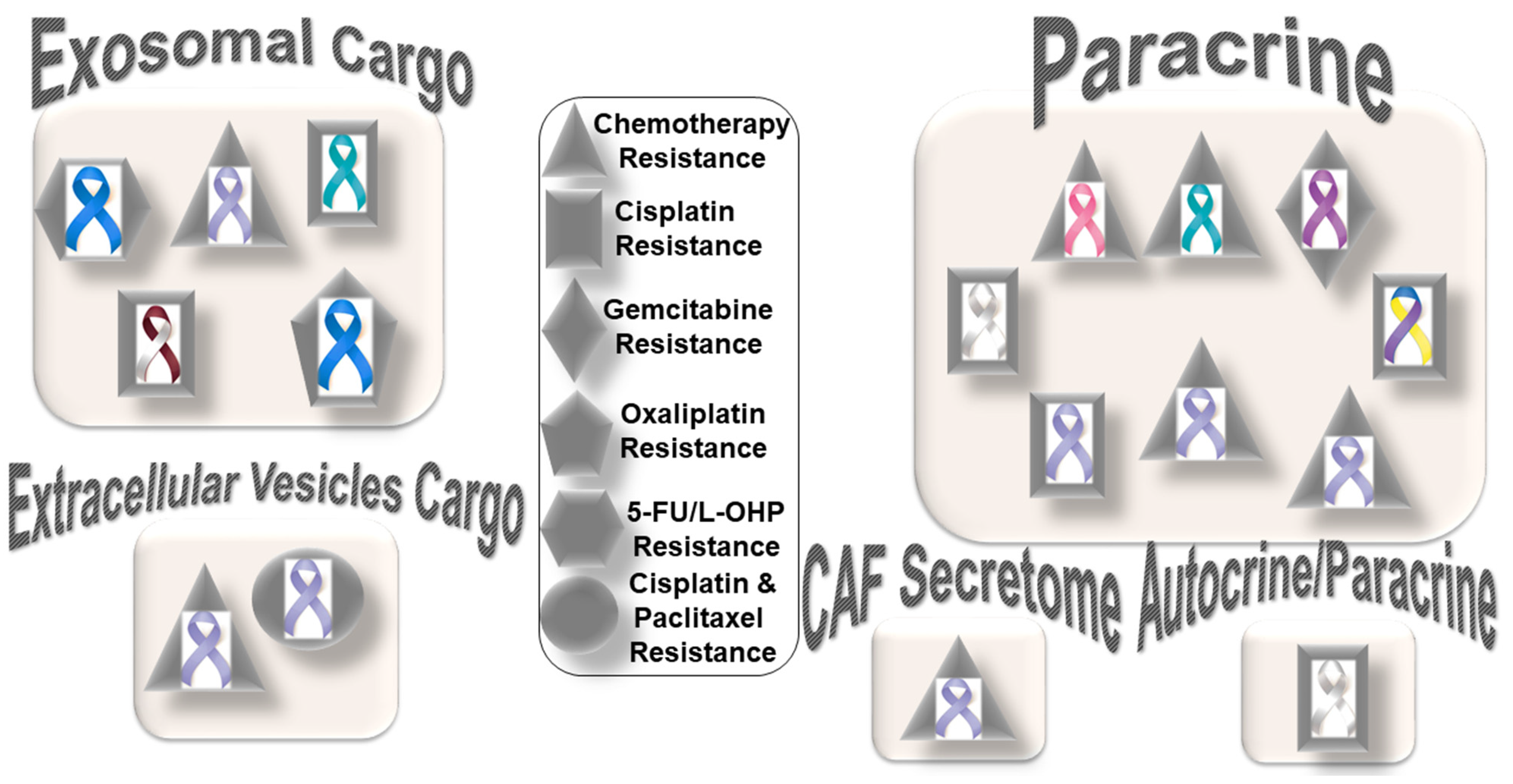
Figure 1. Distribution pattern of types of resistance to chemotherapy based on specific mediators of CAF functions in solid tumors: The four mediators employed by CAFs to orchestrate the development of resistance to chemotherapy are presented in the cartoon. The most common mode of interaction is paracrine, wherein CAFs signal to either tumor cells or other components of the TME via characteristic secretome. In addition to the involvement of the characteristic secretome, exosomal cargos delivering different miRNAs that target various cell signaling proteins are common mediators of CAF actions. Among different organ cancers, gastric cancers have been reported to be the most common tumors in which CAFs are involved in the development of resistance to chemotherapy. The sizes of the boxes indicate the number of studies in each box. The shapes indicate the types of resistance in different tumors (inset). L-OHP is a new derivative of oxaliplatin; 5-FU is fluorouracil. Organ tumors are indicated by their respective ribbon colors. Head and neck cancer: white and burgundy; stomach cancer: periwinkle blue; colon cancer: dark blue; ovarian cancer: teal; lung cancer: white or pearl; breast cancer: pink; pancreatic cancer: purple; bladder cancer: blue, yellow, and purple.
Among the different types of solid tumors, gastric cancers have been reported to be the most common tumors exhibiting CAF-mediated resistance to chemotherapy, which involve paracrine, exosomal cargo, extracellular vesicle, and secretomic modes of action. Secretion of IL-11 from CAFs activated the IL-11/IL-11R/gp130/JAK/STAT3/Bcl anti-apoptosis signaling pathway in gastric cancer cells. Thus, CAF-derived IL-11 secretion caused resistance to chemotherapy regimens in gastric cancers [41]. In another study, CAF-induced activation of the JAK-STAT signaling has been proposed to confer chemoresistance in gastric cancer cells, while interleukin-6 (IL-6) was identified as a CAF-specific secretory protein that protects gastric cancer cells via paracrine signaling. Interestingly, clinical data have shown that IL-6 was differentially expressed in the stromal portion of cancer tissues, while IL-6 upregulation was positively correlated with poor responsiveness to chemotherapy [42]. In line with the above facts, several CAF-targeting agents have been tested in experimental models, as reviewed elsewhere [43]. Resistance to conventional chemotherapeutics in gastric cancers has been reported to be mediated by CAF-derived extracellular vesicles [44]. Annexin A6 initiated network formation and drug resistance within the ECM via activation of beta1 integrin-FAK-YAP signaling. Annexin A6 within CAF extracellular vesicles has been shown to stimulate FAK-YAP signaling by stabilizing beta1 integrin at the cell surface of gastric cancer cells, which subsequently induces drug resistance. In addition to extracellular vesicles, CAFs also communicate via exosomal cargos, which carry miRNAs and mediate resistance to specific chemotherapeutic agents, as presented in the following section.
2.2. CAFs and Specific Resistance to Cisplatin
Reports of CAF-mediated development of cisplatin resistance are more prevalent than any other chemotherapy agent. In certain solid tumors, the mechanism involved intracellular pathway signaling such as JNK or NF-κB, adhesion molecules such as annexin A3, or specific proteins such as plasminogen activator inhibitor-1. In lung cancers, CAFs have been reported to express a higher level of annexin A3 (ANXA3) than normal fibroblasts. The crosstalk was demonstrated using CAF-CM (CAF-conditioned media) incubation, which increased the ANXA3 level in lung cancer cells, which subsequently enhanced cisplatin resistance by inhibiting cisplatin-induced apoptosis involving ANXA3/JNK signaling [45]. In lung adenocarcinoma, cisplatin resistance was associated with the expression of SMAalpha expression [46]. In their study, Masuda et al. demonstrated that the inhibition of plasminogen activator inhibitor-1 increased the chemotherapeutic effect in lung cancer through suppressing the myofibroblast characteristics of CAFs. CAF-derived IL-8 promoted chemoresistance to cisplatin in gastric cancer via NF-κB activation and ABCB1 upregulation [47]. In bladder cancers, stromal CAFs enhanced cisplatin resistance via stimulating IGF-1/ERbeta/Bcl-2 signaling, wherein CAFs regulated ERbeta expression through IGF-1/AKT/c-Jun signaling following c-Jun phosphorylation and promoted ESR2 gene transcription [48]. In other cancers, exosomal cargo carried miRNA to mediate the CAFs’ effect. In ovarian cancer, CAF-mediated cisplatin resistance was reported to involve CAF-derived exosomes, which overexpressed miR-98-5p [49]. In immunocompromised mice, miR-98-5p targeted CDKN1A to inhibit CDKN1A expression and promoted cisplatin resistance by virtue of cell cycle progression. In head and neck cancer, cisplatin resistance is perpetrated by CAF-derived exosomal miR-196a targeting CDKN1B and ING6 [50]. Whether the nature of CAF mediators of cisplatin resistance is organ-specific or not needs to be concluded with more data in this field. From the current literature, it is evident that exosomal miRNA predominantly mediates platinum-based chemotherapy resistance (cisplatin and oxaliplatin), with a few exceptions such as tamoxifen resistance in breast [51] and radioresistance in colorectal cancers [52][53]. In the context of resistance to radiotherapy, CAFs are highly radio-resistant, even at high doses of radiation. CAFs resist apoptosis signals following radiation and become senescent, producing a distinct combination of immunoregulatory molecules. Hence, acquired radio resistance has been associated with CAF function [54][55]. A recent minireview summarized findings on the interactions between CAF, ionizing radiation, and immune cells in the tumor microenvironment [56]. Targeting CAFs, regulatory T cells, and tumor-associated macrophages in combination radio–immunotherapies has been reported to improve cancer treatment [57]. Future studies will also need to clarify the functional segregation of the two modes of events and whether it exists in the development of CAF-mediated resistance in solid tumors.
2.3. CAFs and Specific Resistance to Paclitaxel
CAF-mediated resistance to paclitaxel was reported in ovarian cancers. In ovarian cancers, the lipoma-preferred partner gene has been reported to mediate CAF–endothelial cell crosstalk in signaling chemoresistance [58]. CAFs upregulated the lipoma-preferred partner gene in microvascular endothelial cells via calcium-dependent signaling, and lipoma-preferred partner expression levels in intratumoral microvascular endothelial cells correlated with survival and chemoresistance in patients. Lipoma-preferred partners upregulated focal adhesion and stress fiber formation to promote endothelial cell motility and permeability. Experimental suppression of lipoma-preferred partners improved paclitaxel delivery to cancer cells by decreasing intratumoral microvessel leakiness.
2.4. CAFs and Specific Resistance to a Combination of Cisplatin and Paclitaxel
Specific resistance to a combination of cisplatin and paclitaxel aided by CAFs is encountered in gastric cancers. Exosomal miR-522 suppressed ferroptosis and promoted acquired chemoresistance (decreased chemosensitivity) by targeting ALOX15 and blocking lipid–ROS accumulation involving the intercellular pathway. Both cisplatin and paclitaxel treatment promoted miR-522 secretion from CAFs by activating the USP7/hnRNPA1 axis, leading to ALOX15 suppression and decreased lipid–ROS accumulation in gastric cancer cells [59].
2.5. CAFs and Specific Resistance to Oxaliplatin
CAFs orchestrate oxaliplatin resistance in colorectal cancers [60]. Colorectal cancer-associated lncRNA is transferred from CAFs to the cancer cells via exosomes, where it suppresses colorectal cancer (CRC) cell apoptosis, confers chemoresistance, and activates the Wnt/beta-catenin pathway. Long-non-coding RNA interacts directly with mRNA stabilizing protein (human antigen R) to increase beta-catenin mRNA and protein levels. Specific resistance to 5-FU/L-OHP (oxaliplatin) has been reported in colorectal cancers. In colorectal cancers, chemotherapy resistance was attributed to CAF-secreted exosomes [61]. A direct transfer of exosomes to colorectal tumor cells led to a significant increase in miR-92a-3p levels in cancer cells. An increased expression of miR-92a-3p activated the Wnt/beta-catenin pathway and inhibited mitochondrial apoptosis by directly inhibiting FBXW7 and MOAP1, contributing to stemness, EMT, metastasis, and 5-FU/L-OHP resistance.
2.6. CAFs and Specific Resistance to Gemcitabine
CAF-mediated resistance to gemcitabine involves CAF-derived SDF-1. SDF-1 stimulated malignant progression and gemcitabine resistance in pancreatic cancer due to paracrine induction of SATB-1 within tumor cells. SDF-1-mediated upregulation of SATB-1 expression in tumor cells contributed to the maintenance of CAF properties, forming a reciprocal feedback loop involving the SDF-1/SATB-1 pathway [62]. It is apparent from the results of the above studies that mediators of CAFs in the development of resistance to different chemotherapeutics are specific not only to organ cancers but also the particular drug. In an ideal world, researchers should be searching for an organ-specific blood-based marker that can correlate or indicate CAF-mediated development of resistance to chemotherapy.
3. CAFs and Resistance to Targeted Therapy in Solid Tumors
CAF-mediated resistance to targeted therapy in solid tumors can be categorized into (1) specific resistance to hormone-receptor-targeted anti-cancer drugs and (2) specific resistance to non-hormonal pathway-targeted anti-cancer drugs (Figure 2). One characteristic feature of this type of resistance is the lack of mediation via miRNA compared to resistance to chemotherapy. The only exception to this characteristic is a novel subset of CD63+ CAFs that mediated resistance to tamoxifen in breast cancers via exosomal miR-22 [51]. CD63+ CAFs have been reported to secrete miR-22-rich exosomes, which act through its targets, ERalpha and PTEN, to confer tamoxifen resistance in breast cancer cells. The details of the development of resistance to hormone receptor-targeted anti-cancer drugs mediated by CAFs in breast cancers have been reviewed elsewhere [63]. CAFs have been involved in mediating anti-androgen resistance in prostate cancers in a paracrine manner. Zhang et al. identified neuregulin 1 (NRG1) in the CAF supernatant [64]. CAF-derived NRG1 promoted resistance in tumor cells through the activation of HER3 involving the NRG1/HER3 axis, proving a paracrine mechanism of anti-androgen resistance in prostate cancer. In line with the above fact, an inadequate response to second-generation anti-androgen therapy was recorded in castration-resistant patients with NRG1 activity.
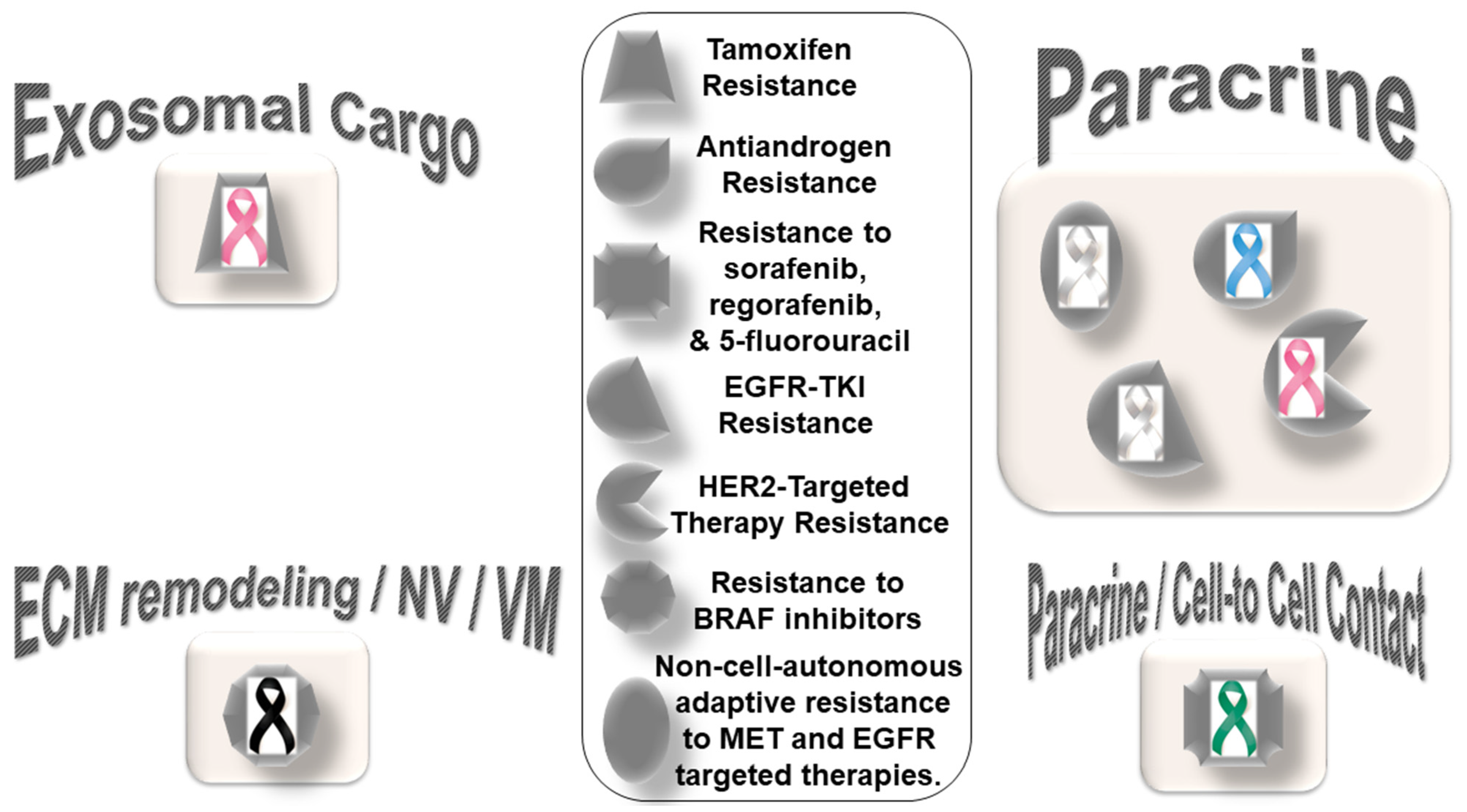
Figure 2. Distribution pattern of types of resistance to targeted therapy based on specific mediators of CAF functions in solid tumors: The four types of mediators of action employed by CAFs to orchestrate the development of resistance to targeted therapy are presented in the cartoon. The most common mode of interaction is paracrine, wherein CAFs signal to either tumor cells or other components of the TME via characteristic secretome. In addition to the involvement of characteristic secretome, exosomal cargos delivering different miRNAs that target various cell signaling proteins are common mediators of CAF action. The sizes of the boxes indicate the number of studies in each box. The shapes indicate the types of resistance in different tumors (inset). Organ tumors are indicated by their respective ribbon colors. Lung cancer: white or pearl; skin cancer: black. liver cancer: emerald green; breast cancer: pink; prostate cancer: light blue.
The role of the activation of EGFR, Wnt/beta-catenin, Hippo, TGF-beta, and JAK/STAT cascades in CAFs in relation to the chemoresistance and invasive or metastatic behavior of cancer cells [65] has strengthened the concept that CAFs should be included as a target for therapy in solid tumors. CAF-mediated resistance to non-hormonal pathway-targeted anti-cancer drugs has been observed in lung, breast, melanoma, and hepatocellular cancers. CAF-mediated non-cell-autonomous adaptive resistance to MET- and EGFR-targeted therapies in lung cancers via a metabolic shift involving paracrine crosstalk between tumor cells under drug exposure and their surrounding CAFs has been reported [66]. Apicella et al. demonstrated that with prolonged exposure to tyrosine kinase inhibitors (TKIs), EGFR- or MET-addicted cancer cells undergo a metabolic shift upregulating glycolysis and lactate production. High secreted levels of lactate stimulate CAFs to produce hepatocyte growth factor (HGF) in a nuclear factor kappa B (NFkB)-dependent manner. This HGF, in turn, activates MET-dependent signaling within cancer cells, counteracting the effects of tyrosine kinase inhibitors (TKIs). In tumor cells of lung adenocarcinoma with EGFR mutations, primary EGFR-TKI resistance was associated with high hepatocyte growth factor in CAFs [67]. Conditioned media from CAFs increased the resistance of PC-9 cells to EGFR-TKI, indicating that with the secretion of higher amounts of CAF-derived humoral factors, HGF is responsible for EGFR-TKI resistance [67]. Understandably, this kind of fail-safe metabolic reprogramming not only allows cellular resistance to the drug but also re-establishes a tumor–TME circuitry, which can also merge with the local immune signaling [68][69][70][71]. As with prostate cancers [64] and melanomas [72], CAFs have been involved in developing resistance to targeted therapies in breast cancers. CAFs participate in the HER2-targeted therapy resistance in breast cancers via the TAF/FGF5/FGFR2/c-Src/HER2 axis [73]. CAF-derived NRG1 (an HER3 ligand) causes resistance to trastuzumab [74][75], TKIs [76], and T-DM1 [77] in HER2-positive breast cancers. In the Neosphere trial, HER2-positive breast tumors with high NRG1 expression appeared to resist trastuzumab–docetaxel but not pertuzumab–trastuzumab–docetaxel [78]. Guardia et al. identified CAFs as the primary source of NRG1 in HER2-positive breast cancers. The study showed their role in mediating resistance to trastuzumab, which can be overcome by dual anti-HER2 blockade following pertuzumab–trastuzumab [78]. Recently, a study examined the value of ‘pathological reactive stroma’ (defined as stromal-predominant breast cancer) as a predictor for trastuzumab resistance in patients with early HER2-positive breast cancer receiving adjuvant therapy in the FinHER phase III trial, reporting an association between trastuzumab resistance and the presence of ‘reactive stroma’ [79]. The pathological reactive stroma and the mRNA gene signatures that reflected reactive stroma were tested in 209 HER2-positive breast cancer samples and were found to be correlated with distant disease-free survival. Interestingly, reactive stroma did not correlate with tumor-infiltrating lymphocytes. The study concluded that the ‘pathological reactive stroma’ in HER2-positive or ER-negative early breast cancer tumors might predict resistance to adjuvant trastuzumab therapy.
In line with the pro-tumorigenic role of ‘pathological reactive stroma’, CAFs are known to promote organoid tumor growth in co-culture. The paracrine crosstalk between CAFs and cancer cells regulated physiological characteristics of CAFs, which in turn imparted resistance to cancer cells. In metastatic melanomas, CAFs resist the function of BRAF inhibitors via their crosstalk with tumor cells (vascular mimicry), the ECM, and endothelial cells (neovascularization). The development of drug resistance to BRAF inhibitors is mediated via ECM reprogramming action of CAFs [19]. Recently, Liu et al. reported the activation of nuclear beta-catenin signaling in melanoma CAFs during the development of resistance to BRAF inhibitor or MEK inhibitors, underscoring the role of BRAF-inhibitor-induced CAF reprogramming in matrix remodeling and the therapeutic escape of melanoma cells [80].
CAF populations expressing FAP/ITGA11/COL1A1/CCN2 have been shown to be negatively correlated with disease-free survival in this cancer. The resistance to BRAF inhibitors is the result of CAF-mediated reprogramming of the ECM. The stiffness of the ECM caused by CAFs has been associated with integrin-dependent signaling. Fibroblast-specific production of CCN2, whose overexpression in melanomas was independent of BRAF mutational status, signals through integrins and was found to be essential for neovascularization and vasculogenic mimicry. In hepatocellular carcinomas, tumor cells resist targeted anti-cancer drugs including sorafenib, regorafenib, and 5-fluorouracil in the presence of CAFs via a direct cell–cell contact, as tested in a transwell system through paracrine signaling [81].
CAF signaling in the development of drug resistance is tumor-specific in prostate cancers and lung adenocarcinomas, as presented above. In prostate cancers, CAF-derived neuregulin 1 NRG1 promotes resistance in tumor cells by activating HER3 involving the NRG1/HER3 axis, proving a paracrine mechanism of antiandrogen resistance in a paracrine manner, as presented above [64]. In lung adenocarcinomas bearing EGFR mutations, primary EGFR-TKI resistance is mediated via hepatocyte growth factor from CAFs. CM from CAFs increased the resistance of EGFR mutant lung adenocarcinoma cell line PC-9 cells to EGFR-TKI, indicating that the secretion of higher amounts of HGF is the robust feature of EGFR-TKI-resistance-promoting CAFs [67]. The mode of action of CAFs and the nature of their involvement with respect to the tumor cells and the TME are less studied. The pattern of crosstalk is just beginning to emerge, which can define distinct therapeutic paradigms. In a recent study, Engelman’s group reported three subtypes of lung CAFs that can influence the personalized treatment of non-small cell lung cancer patients. The 3 subtypes of CAFs identified in their study are (1) subtype I with HGFHigh, FGF7High/Low, p-SMAD2Low, targeting driver, HGF-MET, and FGF7-FGFR2; (2) subtype II with HGFHigh, FGF7High, p-SMAD2Low, targeting driver, and FGF7-FGFR2; and (3) subtype III with HGFLow, FGF7Low, and p-SMAD2High [82]. They reported that specific subtypes are associated with particular functions and clinical responses. Subtype I and II CAFs function to protect cancer cells, while subtype III CAFs are involved with a better clinical response via immune cell migration with additional value in immuno-oncology. In addressing the heterogeneity of CAFs, the study systematically connected functions of subpopulations of lung CAFs to specific functions of CAFs in the context of clinical response and resistance to pathway-targeted drugs. Similar studies in the future will delineate the relationships of the mode of action of CAFs with drugs in organ-type cancers in solid tumors. Despite the different mediating actions of CAFs, it will be imperative to know how CAFs support a tumorigenic pathway in cancer cells in the face of pathway-targeted treatment that ultimately leads to the ineffectiveness of the therapy. Supplemental targeting of CAF signals opens an opportunity to improve personalized medicine and bears the promise of a better outcome.
References
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176.
- Park, D.; Sahai, E.; Rullan, A. SnapShot: Cancer-Associated Fibroblasts. Cell 2020, 181, 486–486.e1.
- De, P.; Aske, J.; Dey, N. Cancer-Associated Fibroblast Functions as a Road-Block in Cancer Therapy. Cancers 2021, 13, 5246.
- De, P.; Aske, J.; Dey, N. Cancer-Associated Fibroblasts in Conversation with Tumor Cells in Endometrial Cancers: A Partner in Crime. Int. J. Mol. Sci. 2021, 22, 9121.
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135.
- Didier Meseure, K.D.A. Cancer Metabolic and immune reprogramming: The intimate interaction between cancer cells and microenviroment. Cancer Prev. Curr. Res. 2014, 1, 21–30.
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001.
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783.
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32.
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e410.
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123.
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548.
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835.
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4820–4833.
- Ji, Z.; Tian, W.; Gao, W.; Zang, R.; Wang, H.; Yang, G. Cancer-Associated Fibroblast-Derived Interleukin-8 Promotes Ovarian Cancer Cell Stemness and Malignancy Through the Notch3-Mediated Signaling. Front. Cell Dev. Biol. 2021, 9, 684505.
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e816.
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55.
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732.
- Leask, A. A centralized communication network: Recent insights into the role of the cancer associated fibroblast in the development of drug resistance in tumors. Semin. Cell Dev. Biol. 2020, 101, 111–114.
- Khan, G.J.; Sun, L.; Khan, S.; Yuan, S.; Nongyue, H. Versatility of Cancer Associated Fibroblasts: Commendable Targets for Anti-tumor Therapy. Curr. Drug Targets 2018, 19, 1573–1588.
- Dzobo, K.; Dandara, C. Architecture of Cancer-Associated Fibroblasts in Tumor Microenvironment: Mapping Their Origins, Heterogeneity, and Role in Cancer Therapy Resistance. Omics. J. Integr. Biol. 2020, 24, 314–339.
- Li, B.; Pei, G.; Yao, J.; Ding, Q.; Jia, P.; Zhao, Z. Cell-type deconvolution analysis identifies cancer-associated myofibroblast component as a poor prognostic factor in multiple cancer types. Oncogene 2021, 40, 4686–4694.
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 3408.
- Miyazaki, Y.; Oda, T.; Mori, N.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts In Vitro. FEBS Open Bio 2020, 10, 2268–2281.
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021, 39, 866–882, Erratum in Cancer Cell 2021, 39, 883.
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351.
- Tran, L.L.; Dang, T.; Thomas, R.; Rowley, D.R. ELF3 mediates IL-1alpha induced differentiation of mesenchymal stem cells to inflammatory iCAFs. Stem Cells 2021, 39, 1766–1777.
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137.
- Pan, X.; Zhou, J.; Xiao, Q.; Fujiwara, K.; Zhang, M.; Mo, G.; Gong, W.; Zheng, L. Cancer-associated fibroblast heterogeneity is associated with organ-specific metastasis in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2021, 14, 184.
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2023–2037.
- Mosa, M.H.; Michels, B.E.; Menche, C.; Nicolas, A.M.; Darvishi, T.; Greten, F.R.; Farin, H.F. A Wnt-Induced Phenotypic Switch in Cancer-Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020, 80, 5569–5582.
- Zeltz, C.; Alam, J.; Liu, H.; Erusappan, P.M.; Hoschuetzky, H.; Molven, A.; Parajuli, H.; Cukierman, E.; Costea, D.E.; Lu, N.; et al. Alpha11beta1 Integrin is Induced in a Subset of Cancer-Associated Fibroblasts in Desmoplastic Tumor Stroma and Mediates In Vitro Cell Migration. Cancers 2019, 11, 765.
- Diaz-Maroto, N.G.; Garcia-Vicien, G.; Polcaro, G.; Banuls, M.; Albert, N.; Villanueva, A.; Mollevi, D.G. The Blockade of Tumoral IL1beta-Mediated Signaling in Normal Colonic Fibroblasts Sensitizes Tumor Cells to Chemotherapy and Prevents Inflammatory CAF Activation. Int. J. Mol. Sci. 2021, 22, 4960.
- de Kruijf, E.M.; van Nes, J.G.; van de Velde, C.J.; Putter, H.; Smit, V.T.; Liefers, G.J.; Kuppen, P.J.; Tollenaar, R.A.; Mesker, W.E. Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast Cancer Res. Treat. 2011, 125, 687–696.
- Lv, Z.; Cai, X.; Weng, X.; Xiao, H.; Du, C.; Cheng, J.; Zhou, L.; Xie, H.; Sun, K.; Wu, J.; et al. Tumor-stroma ratio is a prognostic factor for survival in hepatocellular carcinoma patients after liver resection or transplantation. Surgery 2015, 158, 142–150.
- Mesker, W.E.; Junggeburt, J.M.; Szuhai, K.; de Heer, P.; Morreau, H.; Tanke, H.J.; Tollenaar, R.A. The carcinoma-stromal ratio of colon carcinoma is an independent factor for survival compared to lymph node status and tumor stage. Cell. Oncol. Off. J. Int. Soc. Cell. Oncol. 2007, 29, 387–398.
- Wang, K.; Ma, W.; Wang, J.; Yu, L.; Zhang, X.; Wang, Z.; Tan, B.; Wang, N.; Bai, B.; Yang, S.; et al. Tumor-stroma ratio is an independent predictor for survival in esophageal squamous cell carcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2012, 7, 1457–1461.
- Zhang, T.; Xu, J.; Shen, H.; Dong, W.; Ni, Y.; Du, J. Tumor-stroma ratio is an independent predictor for survival in NSCLC. Int. J. Clin. Exp. Pathol. 2015, 8, 11348–11355.
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.; Wu, J.; et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111.
- Lee, D.; Ham, I.H.; Son, S.Y.; Han, S.U.; Kim, Y.B.; Hur, H. Intratumor stromal proportion predicts aggressive phenotype of gastric signet ring cell carcinomas. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2017, 20, 591–601.
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. 2019, 51, 194–210.
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68.
- Ham, I.H.; Lee, D.; Hur, H. Role of Cancer-Associated Fibroblast in Gastric Cancer Progression and Resistance to Treatments. J. Oncol. 2019, 2019, 6270784.
- Uchihara, T.; Miyake, K.; Yonemura, A.; Komohara, Y.; Itoyama, R.; Koiwa, M.; Yasuda, T.; Arima, K.; Harada, K.; Eto, K.; et al. Extracellular Vesicles from Cancer-Associated Fibroblasts Containing Annexin A6 Induces FAK-YAP Activation by Stabilizing beta1 Integrin, Enhancing Drug Resistance. Cancer Res. 2020, 80, 3222–3235.
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620.
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y.; et al. Inhibition of PAI-1 limits chemotherapy resistance in lung cancer through suppressing myofibroblast characteristics of cancer-associated fibroblasts. J. Cell. Mol. Med. 2019, 23, 2984–2994.
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43.
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERbeta/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375.
- Guo, H.; Ha, C.; Dong, H.; Yang, Z.; Ma, Y.; Ding, Y. Cancer-associated fibroblast-derived exosomal microRNA-98-5p promotes cisplatin resistance in ovarian cancer by targeting CDKN1A. Cancer Cell Int. 2019, 19, 347.
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 12.
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518.
- Chen, X.; Liu, J.; Zhang, Q.; Liu, B.; Cheng, Y.; Zhang, Y.; Sun, Y.; Ge, H.; Liu, Y. Exosome-mediated transfer of miR-93-5p from cancer-associated fibroblasts confer radioresistance in colorectal cancer cells by downregulating FOXA1 and upregulating TGFB3. J. Exp. Clin. Cancer Res. CR 2020, 39, 65.
- Chen, X.; Liu, Y.; Zhang, Q.; Liu, B.; Cheng, Y.; Zhang, Y.; Sun, Y.; Liu, J. Exosomal miR-590-3p derived from cancer-associated fibroblasts confers radioresistance in colorectal cancer. Mol. Therapy Nucleic Acids 2021, 24, 113–126.
- Zhang, H.; Hua, Y.; Jiang, Z.; Yue, J.; Shi, M.; Zhen, X.; Zhang, X.; Yang, L.; Zhou, R.; Wu, S. Cancer-associated Fibroblast-promoted LncRNA DNM3OS Confers Radioresistance by Regulating DNA Damage Response in Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1989–2000.
- Domogauer, J.D.; de Toledo, S.M.; Howell, R.W.; Azzam, E.I. Acquired radioresistance in cancer associated fibroblasts is concomitant with enhanced antioxidant potential and DNA repair capacity. Cell Commun. Signal. CCS 2021, 19, 30.
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635.
- Darragh, L.B.; Oweida, A.J.; Karam, S.D. Overcoming Resistance to Combination Radiation-Immunotherapy: A Focus on Contributing Pathways Within the Tumor Microenvironment. Front. Immunol. 2018, 9, 3154.
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Wong, K.K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2018, 128, 589–606.
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43.
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716.
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91.
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Correction: Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2021, 12, 232.
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of Breast Cancer-Associated Fibroblasts in Tumor Development, Therapy Resistance and Evaluation of Potential Therapeutic Strategies. Curr. Med. Chem. 2018, 25, 3414–3434.
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell 2020, 38, 279–296.e279.
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. CR 2020, 39, 112.
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e846.
- Suzuki, E.; Yamazaki, S.; Naito, T.; Hashimoto, H.; Okubo, S.; Udagawa, H.; Goto, K.; Tsuboi, M.; Ochiai, A.; Ishii, G. Secretion of high amounts of hepatocyte growth factor is a characteristic feature of cancer-associated fibroblasts with EGFR-TKI resistance-promoting phenotype: A study of 18 cases of cancer-associated fibroblasts. Pathol. Int. 2019, 69, 472–480.
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. CR 2015, 34, 111.
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355.
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267.
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451.
- Capparelli, C.; Rosenbaum, S.; Berger, A.C.; Aplin, A.E. Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J. Biol. Chem. 2015, 290, 24267–24277.
- Fernandez-Nogueira, P.; Mancino, M.; Fuster, G.; Lopez-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menendez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-Associated Fibroblasts Promote HER2-Targeted Therapy Resistance through FGFR2 Activation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1432–1448.
- Motoyama, A.B.; Hynes, N.E.; Lane, H.A. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 2002, 62, 3151–3158.
- Watanabe, S.; Yonesaka, K.; Tanizaki, J.; Nonagase, Y.; Takegawa, N.; Haratani, K.; Kawakami, H.; Hayashi, H.; Takeda, M.; Tsurutani, J.; et al. Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Med. 2019, 8, 1258–1268.
- Xia, W.; Petricoin, E.F., 3rd; Zhao, S.; Liu, L.; Osada, T.; Cheng, Q.; Wulfkuhle, J.D.; Gwin, W.R.; Yang, X.; Gallagher, R.I.; et al. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res. BCR 2013, 15, R85.
- Phillips, G.D.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A., 3rd; Albain, K.S.; Harbeck, N.; et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: Critical role for neuregulin blockade in antitumor response to combination therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 456–468.
- Guardia, C.; Bianchini, G.; Arpi, L.O.; Menendez, S.; Casadevall, D.; Galbardi, B.; Dugo, M.; Servitja, S.; Montero, J.C.; Soria-Jimenez, L.; et al. Preclinical and Clinical Characterization of Fibroblast-derived Neuregulin-1 on Trastuzumab and Pertuzumab Activity in HER2-positive Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5096–5108.
- Sonnenblick, A.; Salmon-Divon, M.; Salgado, R.; Dvash, E.; Ponde, N.; Zahavi, T.; Salmon, A.; Loibl, S.; Denkert, C.; Joensuu, H.; et al. Reactive stroma and trastuzumab resistance in HER2-positive early breast cancer. Int. J. Cancer 2020, 147, 266–276.
- Liu, T.; Zhou, L.; Xiao, Y.; Andl, T.; Zhang, Y. BRAF Inhibitors Reprogram Cancer-Associated Fibroblasts to Drive Matrix Remodeling and Therapeutic Escape in Melanoma. Cancer Res. 2022, 82, 419–432.
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.A.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431.
- Hu, H.; Piotrowska, Z.; Hare, P.J.; Chen, H.; Mulvey, H.E.; Mayfield, A.; Noeen, S.; Kattermann, K.; Greenberg, M.; Williams, A.; et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell 2021, 11, 1531–1547.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
545
Revisions:
2 times
(View History)
Update Date:
31 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No