Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Federica Cherchi | -- | 1778 | 2022-03-31 10:56:43 | | | |
| 2 | Vicky Zhou | Meta information modification | 1778 | 2022-04-06 11:03:33 | | | | |
| 3 | Vicky Zhou | Meta information modification | 1778 | 2022-04-06 11:06:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cherchi, F.; Coppi, E.; Venturini, M.; Lucarini, E.; Corradetti, R.; Di Cesare Mannelli, L.; Ghelardini, C.; Pedata, F.; Pugliese, A.M. Ligands of the Gi Protein-Coupled Adenosine A3 Receptor. Encyclopedia. Available online: https://encyclopedia.pub/entry/21206 (accessed on 10 August 2026).
Cherchi F, Coppi E, Venturini M, Lucarini E, Corradetti R, Di Cesare Mannelli L, et al. Ligands of the Gi Protein-Coupled Adenosine A3 Receptor. Encyclopedia. Available at: https://encyclopedia.pub/entry/21206. Accessed August 10, 2026.
Cherchi, Federica, Elisabetta Coppi, Martina Venturini, Elena Lucarini, Renato Corradetti, Lorenzo Di Cesare Mannelli, Carla Ghelardini, Felicita Pedata, Anna Maria Pugliese. "Ligands of the Gi Protein-Coupled Adenosine A3 Receptor" Encyclopedia, https://encyclopedia.pub/entry/21206 (accessed August 10, 2026).
Cherchi, F., Coppi, E., Venturini, M., Lucarini, E., Corradetti, R., Di Cesare Mannelli, L., Ghelardini, C., Pedata, F., & Pugliese, A.M. (2022, March 31). Ligands of the Gi Protein-Coupled Adenosine A3 Receptor. In Encyclopedia. https://encyclopedia.pub/entry/21206
Cherchi, Federica, et al. "Ligands of the Gi Protein-Coupled Adenosine A3 Receptor." Encyclopedia. Web. 31 March, 2022.
Copy Citation
Ligands of the Gi protein-coupled adenosine A3 receptor (A3R) are receiving increasing interest as attractive therapeutic tools for the treatment of a number of pathological conditions of the central and peripheral nervous systems (CNS and PNS, respectively). Their safe pharmacological profiles emerging from clinical trials on different pathologies (e.g., rheumatoid arthritis, psoriasis and fatty liver diseases) confer a realistic translational potential to these compounds, thus encouraging the investigation of highly selective agonists and antagonists of A3R.
adenosine receptors
Adenosine A3 ligands
chronic pain
brain ischemia
1. Introduction: Adenosine and Its Receptors
Adenosine is a ubiquitous endogenous neuromodulator and is recognized as one of the most evolutionarily ancient and pervasive signaling molecules [1]. In particular, adenosine plays a crucial role in neuron-to-glia communication in both the central and peripheral nervous systems (CNS and PNS, respectively) [2][3][4] as well as in inflammatory processes throughout the periphery [5]. The effects of adenosine are mediated by the activation of four G protein-coupled receptors (GPCR): A1, A2A, A2B and A3 receptors (A1Rs, A2ARs, A2BRs and A3Rs). A1Rs, A2ARs and A2BRs are fairly conserved throughout evolution, showing 80–95% homology among species, whereas A3Rs present high inter-species variability [6], with only 74% sequence homology between rat and human [7]. In general, A1Rs and A3Rs are coupled to adenylyl cyclase (AC) inhibition through G proteins of the Gi and Go family [8][9] (Figure 1), whereas A2AR and A2BR actions rely on the stimulation of adenylyl cyclase by Gs or Golf. However, beyond these canonical signaling mechanisms, the stimulation of A1Rs and A3Rs can also elicit Gq-mediated release of calcium ions from intracellular stores [10]. On the other hand, in addition to Gs, A2BR receptors can activate phospholipase C, which also occurs through Gq [10] (Figure 1). Moreover, all adenosine receptors are coupled to mitogen-activated protein kinase (MAPK) pathways, which include extracellular signal-regulated kinase 1 (ERK1), ERK2, JUN N-terminal kinase and p38 MAPK [10].
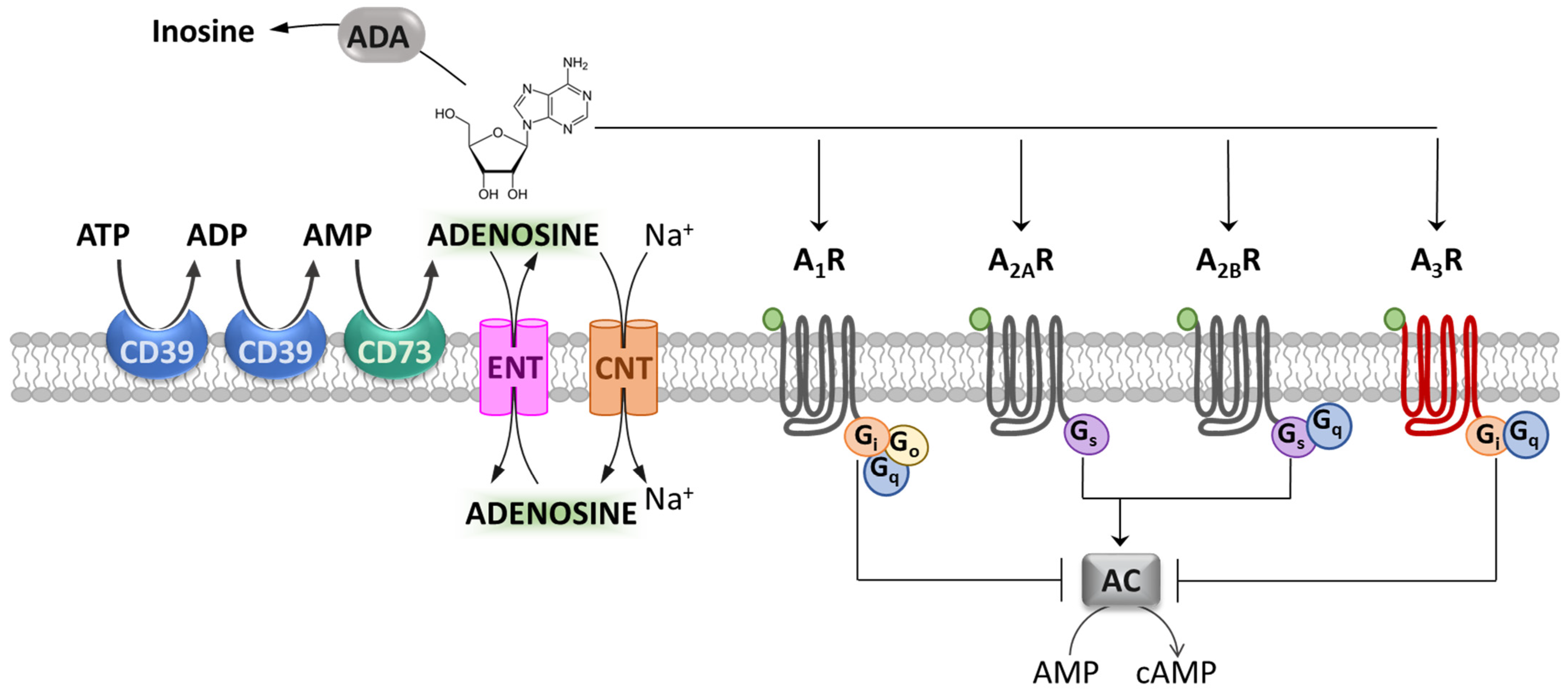
Figure 1. Adenosine and its receptors. Schematic representation of adenosine metabolism, transport and metabotropic receptors. The ectonucleotidases CD39 and CD73 metabolize ATP and ADP to AMP, and AMP to adenosine. The equilibrative nucleoside transporter (ENT) or concentrative nucleoside transporter (CNT) families mediate adenosine reuptake. Adenosine metabotropic receptors (A1, A2A, A2B and A3 receptors: A1R, A2AR, A2BR and A3R) are differently coupled to adenylyl cyclase (AC) inhibition or stimulation. Adenosine deaminase (ADA) deactivates extracellular adenosine by converting it into inosine.
The activation of each adenosine receptor subtype depends upon extracellular adenosine levels, which, in turn, are regulated by a number of intra- or extracellular adenosine-synthetizing and degrading enzymes, as well as an uptake system at the membrane level. This complex machinery allows tissues to modulate purinergic signaling according to variations in tissue health status [11]. In the extracellular space, the ectonucleotidases CD39 and CD73 metabolize ATP and ADP to AMP, and AMP to adenosine, respectively, and they are the major sources of extracellular adenosine [12]. On the other hand, adenosine is removed by enzymes dedicated to its degradation, such as adenosine deaminase (ADA), which degrades extracellular adenosine to inosine (Figure 1). These catabolic enzymes, together with equilibrative nucleoside transporters (ENTs) and concentrative nucleoside transporters (CNTs) on the cell membrane, shunt extracellular adenosine into the intracellular space, thereby terminating adenosine receptor signaling [13][14].
Under physiological conditions, endogenous adenosine levels fall into the nanomolar range in the vast majority of organs and tissues [15]. However, during trauma or injury, particularly during hypoxic/ischemic insults [16][17][18], adenosine is released in massive quantities by damaged cells and reaches micromolar concentrations that can activate all subtypes of adenosine receptors [11].
1.1. Adenosine Receptors in Peripheral Tissues
In the periphery, adenosine receptors modulate a number of events, including inflammation, metabolism and cell-to-cell signaling. A pervasive action of peripheral adenosine is on A1Rs in the heart (Figure 2), where they are highly expressed and mediate potent bradycardia. For this reason, adenosine is administered as an emergency drug in arrhythmic conditions, e.g., paroxysmal supraventricular tachycardia [19].
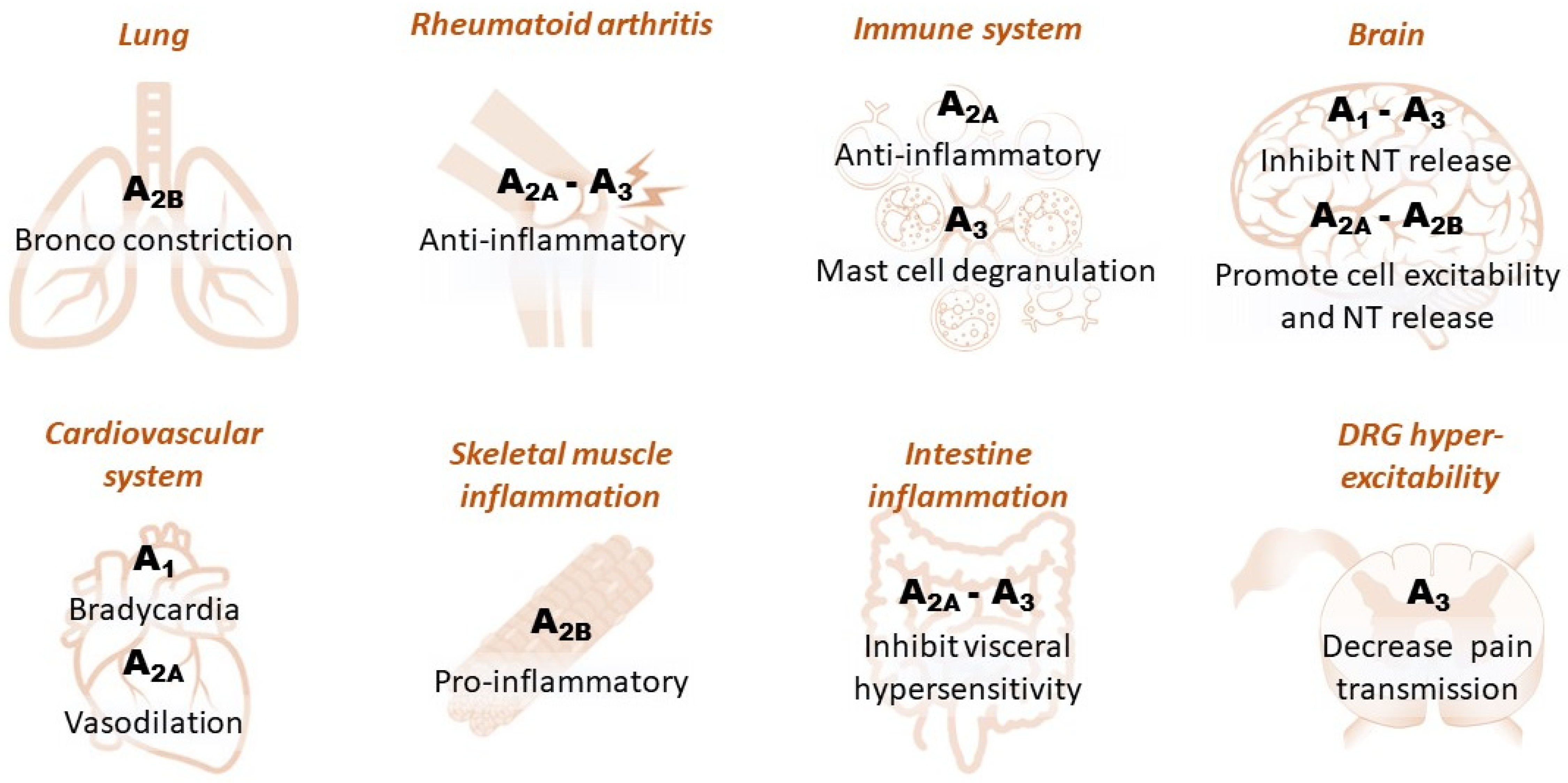
Figure 2. Schematic diagram illustrating the effect of different adenosine receptor subtypes in peripheral and central tissues. Dorsal root ganglia: DRG.
1.2. Adenosine Receptors in the CNS
At the central level, A1Rs are the primary effectors of adenosine actions in the CNS, where they are widely distributed, with the highest levels reported in the cerebral cortex and hippocampus [20]. A1Rs are known to inhibit neurotransmitter release by inhibiting presynaptic voltage-gated calcium channels (VGCCs) [21][22] and to decrease neuronal excitability by increasing postsynaptic potassium conductance [23]. During brain ischemia, these actions provide important adenosine-mediated neuroprotection by maintaining cells in a low energy consumption state and by counteracting excessive glutamate release, which is responsible for the excitotoxic damage [24].
The functional effect of the Gs-coupled A2AR subtype in the CNS is the opposite to that of A1Rs, as these receptors are reported to enhance glutamate release by facilitating calcium entry through presynaptic VGCCs and inhibiting its uptake [25]. Moreover, A2AR inhibits voltage-dependent potassium channels, thus promoting cell excitability and neurotransmitter release [26] and exacerbating excitotoxic damage during ischemic/hypoxic conditions [26]. The expression levels of A2AR in the CNS are comparable to those of A1Rs only in the caudate/putamen nuclei, where they play a deleterious role in the acute phases of an ischemic insult by increasing brain damage and neurological deficit [27] after middle cerebral artery occlusion (MCAo) in rats [16].
2. Therapeutic Potential of A3R Ligands
2.1. A3R Ligands as Therapeutic Tools in Brain Ischemia
There are no current treatments for stroke that are able to prevent or alleviate neuronal loss and tissue damage in the acute or post-ischemic phases after the insult. The only therapy strategies currently in use are the administration of tissue plasminogen activator (tPA) within the first 3–4 h after the insult [28] and, in some cases, hypothermia [29][30]. As stated above, adenosine is released in great amounts during hypoxic/ischemic conditions and plays a pervasive role in this pathology by activating all subtypes of P1 receptors. Both protective and deleterious effects have been described upon their selective activation, depending on the receptor subtype and timing after the insult.
The most widely recognized effect of adenosine during brain ischemia is the neuroprotective role exerted by the Gi-coupled A1R subtype, which is known to inhibit exaggerated glutamate release during excitotoxic damage in the ischemic core and penumbra [22][23]. As a result of the wide distribution of A1Rs within the brain, they have a higher impact compared to A2AR, A2BR and A3R subtypes [18]. Unfortunately, due to unacceptable side effects at the peripheral level (bradycardia and hypotension), A1R-selective agonists are not suitable for clinical use in preventing post-ischemic damage [23].
Hence, research was driven towards the other Gi-coupled adenosine receptor subtype, A3R. Interestingly, A3R has been proposed as a novel therapeutic target for a number of pathologies [31][32], and A3R-selective nucleosides are already in phase 2 and/or 3 clinical trials for autoimmune inflammatory diseases, liver cancer and non-alcoholic steatohepatitis (see www.clinicaltrials.gov; NCT00556894; NCT02927314; 12 March 2022) [33][34]. Notably, activation of A3R in humans by the selective and orally bioavailable A3R agonist IB-MECA (1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purine-9-yl]-N-methyl-β-D-ribofuranuronamide) and its chlorinated counterpart Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)-adenosine-5-N-methyluronamide) is not associated with cardiac or hemodynamic effects [34], and they have shown encouragingly safe profiles [33]. Further attention was focused on this adenosine receptor subtype for the investigation of innovative pain-relieving strategies, as detailed below, due to evidence for their good “safety and security” profile.
2.2. A3R Ligands as Therapeutic Tools in Chronic Pain
Chronic pain is a highly debilitating condition, disturbing all aspects of people's daily experience, from social life to career-related contexts. The pharmacological tools available to date are sometimes inadequate or, as in the case of opioids, limited by serious adverse effects [35]. In an effort to find innovative, non-opioid pain-relieving compounds, many experimental reports have identified adenosine receptors as potential targets for acute or chronic pain management.
The first proof of adenosine’s anti-nociceptive effect dates from the 1970s, when administration of adenosinergic agonists proved effective in pain control. These studies emphasized the role of adenosine A1Rs in producing anti-nociceptive effects, with some effects ascribed to the A2AR subtype [36][37]. Adenosine involvement in peripheral nociception was further confirmed; e.g., the local administration of exogenous A1R agonists to the hind paw of the rat produces anti-nociceptive effects in a pressure hyperalgesia model [38], whereas local administration of A2R agonists enhances pain responses [39], an action due to adenosine A2AR activation, as confirmed by using the selective agonist CGS21680 [40]. Later, it was demonstrated that the anti-nociceptive action of A1R agonists could be ascribed to AC inhibition and to the consequent decrease in cAMP production in sensory nerve terminals [41][42]; thus, a robust protective role of A1R agonists emerged [43]. On the other hand, the A2AR-mediated promotion of cutaneous pain resulted from the stimulation of AC, leading to increased cAMP levels in sensory nerve terminals [41][42], thus producing opposite effects to those elicited by the anti-hyperalgesic, Gi-coupled A1R subtype. However, the relation between A2ARs and pain has been controversial, with evidence supporting either pro-nociceptive or anti-nociceptive activity depending on the receptor localization and animal models of pain [43]. Indeed, a relevant A2AR-mediated anti-nociceptive effect was described in a recent study demonstrating that central neuropathic pain evoked by dorsal root avulsion could be reversed by a single intrathecal injection of A2AR agonists [44].
Importantly, the effect of MRS5980 on neuronal firing was also abolished by pre-treatment with an anti-IL-10-selective antibody, thus unequivocally pointing to this anti-inflammatory cytokine as the main effector produced by CD4+ T cells upon A3R activation to inhibit neuronal excitability (Figure 3) [45].
 Figure 3. Adenosine A3 receptors and pain control. A3 receptors (A3Rs) are expressed on rat DRG neurons, and their activation by the selective agonist MRS5980 decreases action potential (AP) neuronal firing and inhibits N-type voltage-gated calcium channels [46]. A3Rs expressed on CD4+ T cells, but not on mouse DRG neurons, promote interleukin-10 (IL-10) release that, by activating IL-10 receptors (IL-10R) on DRG neurons, reduces neuronal excitability by inhibiting AP firing [45].
Figure 3. Adenosine A3 receptors and pain control. A3 receptors (A3Rs) are expressed on rat DRG neurons, and their activation by the selective agonist MRS5980 decreases action potential (AP) neuronal firing and inhibits N-type voltage-gated calcium channels [46]. A3Rs expressed on CD4+ T cells, but not on mouse DRG neurons, promote interleukin-10 (IL-10) release that, by activating IL-10 receptors (IL-10R) on DRG neurons, reduces neuronal excitability by inhibiting AP firing [45].3. Conclusions
In summary, it can be concluded that A3Rs are emerging as promising targets for the treatment of a number of pathologies due to the limited side effects of their ligands in comparison to those targeting other adenosine receptor subtypes (i.e., A1Rs or A2ARs). In particular, A3R antagonists proved effective in preclinical animal models of brain ischemia and OGD in hippocampal slices, thus paving the way for the development of new, highly selective A3R blockers for the treatment of stroke. Furthermore, valuable evidence from rodent models of chronic pain indicates the possible use of selective A3R agonists as non-narcotic anti-hyperalgesic agents for pain control.
References
- Verkhratsky, A. Early evolutionary history (from bacteria to hemichordata) of the omnipresent purinergic signalling: A tribute to Geoff Burnstock inquisitive mind. Biochem. Pharmacol. 2021, 187, 114261.
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J. Neurophysiol. 2012, 107, 1009–1021.
- Coppi, E.; Cellai, L.; Maraula, G.; Dettori, I.; Melani, A.; Pugliese, A.M.; Pedata, F. Role of adenosine in oligodendrocyte precursor maturation. Front. Cell. Neurosci. 2015, 9, 155.
- Verkhrasky, A.; Krishtal, O.A.; Burnstock, G. Purinoceptors on neuroglia. Mol. Neurobiol. 2009, 39, 190–208.
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857.
- Linden, J. Cloned adenosine A3 receptors: Pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306.
- Koscsó, B.; Csóka, B.; Pacher, P.; Haskó, G. Investigational A3 adenosine receptor targeting agents. Expert Opin. Investig. Drugs 2011, 20, 757–768.
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34.
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell. Signal. 2002, 14, 99–108.
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535.
- Antonioli, L.; Colucci, R.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr. Drug Targets 2012, 13, 842–862.
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367.
- Baldwin, S.A.; Beal, P.R.; Yao, S.Y.M.; King, A.E.; Cass, C.E.; Young, J.D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004, 447, 735–743.
- Gray, J.H.; Owen, R.P.; Giacomini, K.M. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. 2004, 447, 728–734.
- Pedata, F.; Corsi, C.; Melani, A.; Bordoni, F.; Latini, S. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann. N. Y. Acad. Sci. 2001, 939, 74–84.
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130.
- Fredholm, B.B.; Dunwiddie, T.V.; Bergman, B.; Lindström, K. Levels of adenosine and adenine nucleotides in slices of rat hippocampus. Brain Res. 1984, 295, 127–136.
- Dunwiddie, T.V.; Diao, L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J. Pharmacol. Exp. Ther. 1994, 268, 537–545.
- Al-Zaiti, S.S.; Magdic, K.S. Paroxysmal Supraventricular Tachycardia: Pathophysiology, Diagnosis, and Management. Crit. Care Nurs. Clin. N. Am. 2016, 28, 309–316.
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.S.; Richardson, P.J.; Freeman, T.C. Tissue distribution of adenosine receptor mRNAs in the rat. Br. J. Pharmacol. 1996, 118, 1461–1468.
- Andiné, P. Involvement of adenosine in ischemic and postischemic calcium regulation. Mol. Chem. Neuropathol. 1993, 18, 35–49.
- Corradetti, R.; Lo Conte, G.; Moroni, F.; Beatrice Passani, M.; Pepeu, G. Adenosine decreases aspartate and glutamate release from rat hippocampal slices. Eur. J. Pharmacol. 1984, 104, 19–26.
- Deb, P.K.; Deka, S.; Borah, P.; Abed, S.N.; Klotz, K.-N. Medicinal Chemistry and Therapeutic Potential of Agonists, Antagonists and Allosteric Modulators of A1 Adenosine Receptor: Current Status and Perspectives. Curr. Pharm. Des. 2019, 25, 2697–2715.
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321.
- Gonçalves, M.L.; Ribeiro, J.A. Adenosine A2 receptor activation facilitates 45Ca2+ uptake by rat brain synaptosomes. Eur. J. Pharmacol. 1996, 310, 257–261.
- Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. Adenosine A2A receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1 receptor inhibition. Neuroscience 2002, 112, 319–329.
- Melani, A.; Gianfriddo, M.; Vannucchi, M.G.; Cipriani, S.; Baraldi, P.G.; Giovannini, M.G.; Pedata, F. The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006, 1073–1074, 470–480.
- Kleindorfer, D.; Lindsell, C.J.; Brass, L.; Koroshetz, W.; Broderick, J.P. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke 2008, 39, 924–928.
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278.
- Diller, K.R.; Zhu, L. Hypothermia Therapy for Brain Injury. Annu. Rev. Biomed. Eng. 2009, 11, 135–162.
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A 3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366.
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264.
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell. Neurosci. 2019, 13, 124.
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J. Rheumatol. 2008, 35, 41–48.
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet. Neurol. 2015, 14, 162–173.
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338, 1–18.
- Yoon, M.H.; Bae, H.B.; Choi, J. Il Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth. Analg. 2005, 101, 1417–1421.
- Taiwo, Y.O.; Levine, J.D. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 1990, 38, 757–762.
- Karlsten, R.; Gordh, T.; Post, C. Local Antinociceptive and Hyper algesic Effects in the Formalin Test after Peripheral Administration of Adenosine Analogues in Mice. Pharmacol. Toxicol. 1992, 70, 434–438.
- Doak, G.J.; Sawynok, J. Complex role of peripheral adenosine in the genesis of the response to subcutaneous formalin in the rat. Eur. J. Pharmacol. 1995, 281, 311–318.
- Taiwo, Y.O.; Levine, J.D. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience 1991, 44, 131–135.
- Khasar, S.G.; Wang, J.F.; Taiwo, Y.O.; Heller, P.H.; Green, P.G.; Levine, J.D. Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat: A G-protein βγ subunit-mediated effect? Neuroscience 1995, 67, 189–195.
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196.
- Kwilasz, A.J.; Ellis, A.; Wieseler, J.; Loram, L.; Favret, J.; McFadden, A.; Springer, K.; Falci, S.; Rieger, J.; Maier, S.F.; et al. Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A 2A receptor agonists. Brain. Behav. Immun. 2018, 69, 470–479.
- Durante, M.; Squillace, S.; Lauro, F.; Giancotti, L.A.; Coppi, E.; Cherchi, F.; Di Cesare Mannelli, L.; Ghelardini, C.; Kolar, G.; Wahlman, C.; et al. Adenosine A3 agonists reverse neuropathic pain via T cell–mediated production of IL-10. J. Clin. Investig. 2021, 131.
- Jain, K.K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert Opin. Investig. Drugs 2000, 9, 2403–2410.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
3 times
(View History)
Update Date:
06 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No