Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robert Brown | + 2375 word(s) | 2375 | 2022-03-21 04:58:53 | | | |
| 2 | Camila Xu | Meta information modification | 2375 | 2022-03-28 08:17:49 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Brown, R. Epigenetic Therapies in Modulating DNA Damage Repair Pathways. Encyclopedia. Available online: https://encyclopedia.pub/entry/21069 (accessed on 01 July 2026).
Brown R. Epigenetic Therapies in Modulating DNA Damage Repair Pathways. Encyclopedia. Available at: https://encyclopedia.pub/entry/21069. Accessed July 01, 2026.
Brown, Robert. "Epigenetic Therapies in Modulating DNA Damage Repair Pathways" Encyclopedia, https://encyclopedia.pub/entry/21069 (accessed July 01, 2026).
Brown, R. (2022, March 25). Epigenetic Therapies in Modulating DNA Damage Repair Pathways. In Encyclopedia. https://encyclopedia.pub/entry/21069
Brown, Robert. "Epigenetic Therapies in Modulating DNA Damage Repair Pathways." Encyclopedia. Web. 25 March, 2022.
Copy Citation
Epigenetic therapies describe drug molecules such as DNA methyltransferase, histone methyltransferase and histone acetylase/deacetylase inhibitors, which target epigenetic mechanisms such as DNA methylation and histone modifications. Many DNA damage response (DDR) genes are epigenetically regulated in cancer leading to transcriptional silencing and the loss of DNA repair capacity. Epigenetic marks at DDR genes, such as DNA methylation at gene promoters, have the potential to be used as stratification biomarkers, identifying which patients may benefit from particular chemotherapy treatments.
DNA repair
epigenetics
cancer
DNA methylation
1. Deoxyribonucleic Acid (DNA) Damage Response (DDR) Genes Are Epigenetically Regulated in Cancer, Affecting Chemosensitivity
Epigenetic modifications, such as gene promoter DNA hypermethylation, and subsequent changes in gene expression of DDR-associated genes, lead to a loss of DNA repair capacity and have been demonstrated in a variety of tumours and cell line models [1][2]. The loss of DNA repair activity in tumours may lead to chemosensitivity to DNA-damaging cytotoxic chemotherapy. One of the paradigms demonstrating the clinical relevance of epigenetic mechanisms involved in chemosensitivity is highlighted in glioblastoma. DNA methylation of MGMT, the DNA repair gene encoding O-6-methylguanine-DNA methyltransferase, leads to the loss of MGMT expression [1]. This leads to reduced DNA damage repair and subsequently increased sensitivity of cells to the alkylating agent, temozolomide [3]. Early clinical studies showed that glioma patients treated with temozolomide and radiotherapy and with a methylated MGMT gene promoter have a survival benefit compared to only radiotherapy. Patients with unmethylated MGMT promoters showed no statistically significant difference in survival [4]. Prospective randomised trials of glioblastoma patients for radiotherapy versus alkylating agent chemotherapy have demonstrated DNA methylation of MGMT is a clinically useful predictive biomarker to stratify patients, rather than just prognostic [3][5][6].
A second paradigm is the epigenetic alterations of homology recombination DNA repair (HR)-associated genes including breast cancer type 2 susceptibility proteins 1 and 2 (BRCA1/2) [7][8]. BRCA1 is frequently methylated in high-grade serous ovarian cancer (HGSOC) and can lead to HR-deficiency (HRD) which is associated with increased patient survival following platinum-based chemotherapy compared to patients with HR proficient tumours [8]. Sensitivity to platinum-based chemotherapy exploits HRD in HGSOC by introducing double-stranded breaks in DNA, leading to genomic instability and cell apoptosis [9]. However, non-homologous end joining (NHEJ) and base excision repair (BER) pathways, which require Poly (ADP-ribose) polymerase (PARP), can still be utilised to repair the damaged DNA. PARP inhibitors (PARPi) induce synthetic lethality in HR-deficient tumours by disrupting multiple DNA repair pathways simultaneously [10]. In breast and ovarian cancer, BRCA1/2 status is clinically useful to predict sensitivity to platinum-based chemotherapy and PARPi [11][12]. Furthermore, patients with BRCA1 methylated and HRD HGSOC have better prognosis than unmethylated HR proficient tumours [13].
In both the above paradigms, epigenetic regulation by DNA methylation during tumour development, prior to chemotherapy, leads to inactivation of DNA repair activity and drug sensitivity. However, epigenetic mechanisms have also been proposed as important drivers of acquired drug resistance adaptation during chemotherapy. This leads to increased epigenetic silencing in tumours at relapse compared to primary presentation [14]. For instance, loss of DNA mismatch repair (MMR) due to DNA methylation at the MutL Homolog 1 (MLH1) gene promoter has been associated with resistance to alkylating agents such as temozolomide and crosslinking agents such as cisplatin [15][16][17]. The presence of functional MMR has been proposed to lead to cell death due to futile repair cycles, generation of double-strand DNA breaks and engagement of apoptosis [18]. Thus, the absence of MMR leads to loss of engagement of cell death pathways by DDR pathways leading to drug resistance. In another example of epigenetic adaptation following chemotherapy, while sensitivity to PARP inhibitors of HGSOC is associated with DNA methylation at BRCA1, tumours recurring following chemotherapy restore BRCA1 expression associated with reduced DNA methylation. This supports a key role for DNA methylation changes during the acquisition of PARPi resistance [19]. FANCF, another DDR-associated gene closely linked to BRCA genes, is often methylated in several different cancer types including testicular [20], head and neck [21], lung [21], cervical [22] and ovarian [23][24]. Methylation of the FANCF promoter in ovarian cancer has been linked to platinum sensitivity, whereas demethylation of FANCF has been associated with platinum resistance and often occurs after platinum chemotherapy [24][25].
Whilst aberrant methylation of DDR genes has been shown in multiple cancers, other epigenetic mechanisms such as histone modifications at genomic regulatory regions, including enhancers and super enhancers, may also play an important role in response to chemotherapy. Studies show modest benefits of temozolmide treatment in patients with methylated MGMT in colorectal cancer and patient-derived xenograft (PDX) models of glioblastoma show high expression of MGMT linked to active enhancers, despite promoter methylation [26]. This suggests different epigenetic mechanisms are able to dynamically regulate gene expression. Furthermore, DDR-associated genes associated with drug response may themselves regulate the epigenetic landscape. BRCA1 mutations in breast cancer epithelial cells lead to the loss of H3K27ac at super enhancers and impair enhancer–promoter lopping [27], whereas BRCA2 depletion has been linked to chromatin remodelling [28]. MMR inactivation via MLH1 mutations has been shown to activate enhancers of genes associated with growth in colorectal cancer and may activate enhancers of genes associated with drug-resistance [29]. These observations highlight the potential interplay of different epigenetic mechanisms and DDR-associated genes in relation to chemosensitivity (Figure 1).
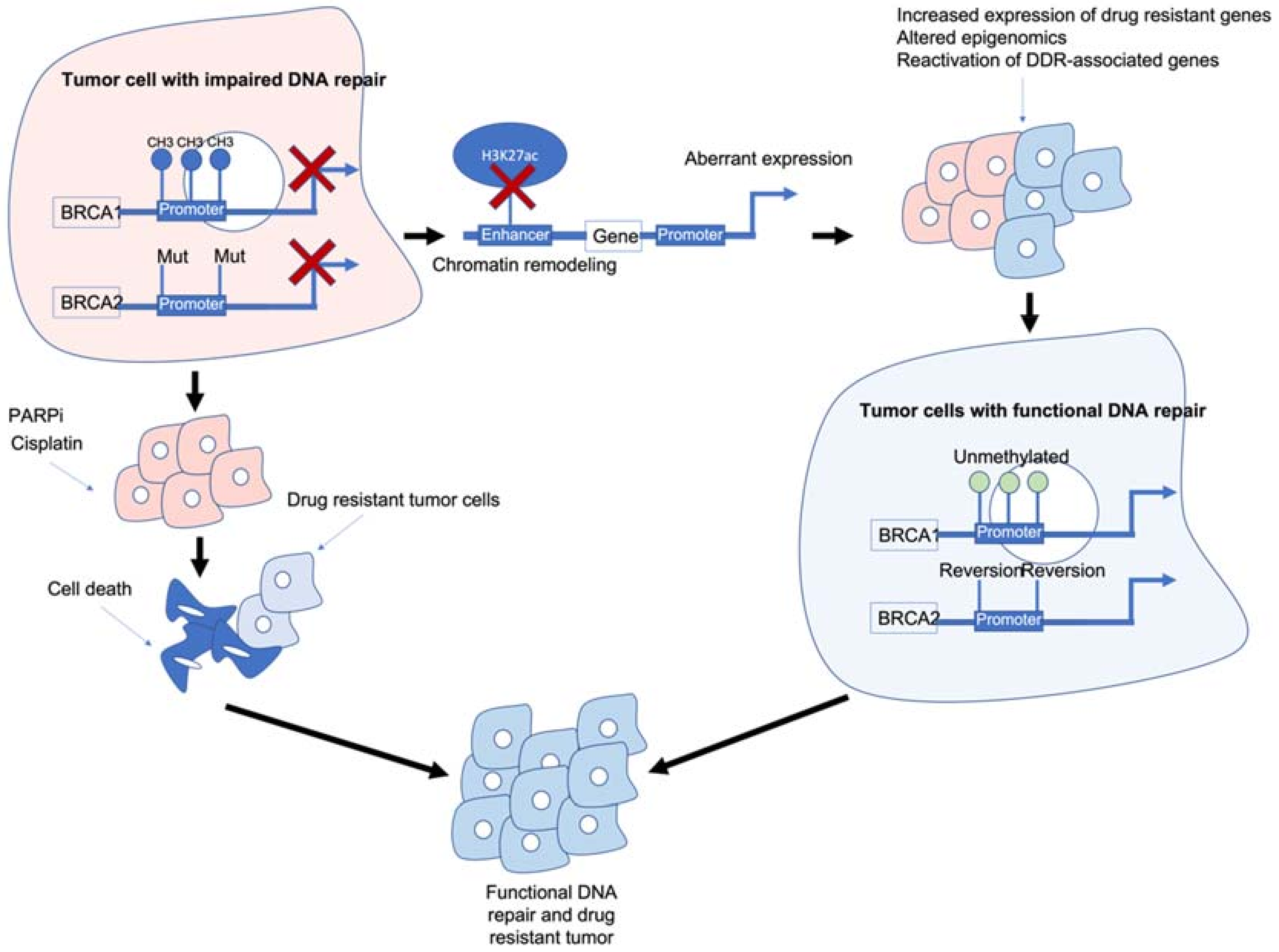 Figure 1. Epigenetic interactions with breast cancer type 1/2 susceptibility (BRCA1/2) genes and drug resistance. BRCA1/2 deficiency caused by methylation and/or mutations results in impaired DNA repair and often sensitivity to poly (ADP-ribose) polymerase inhibitors (PARPi)/platinum-based chemotherapies. BRCA1/2 deficiency can also modulate chromatin accessibility and enhancers of drug-resistant and/or DNA damage response (DDR)-associated genes. Reactivation of BRCA1/2 either by demethylation or reversion mutations can result in tumour cells with functional DDR. Ultimately, these mechanisms result in tumour cells that are drug resistant, have functional DDR and no longer respond to previous PARPi/platinum chemotherapy.
Figure 1. Epigenetic interactions with breast cancer type 1/2 susceptibility (BRCA1/2) genes and drug resistance. BRCA1/2 deficiency caused by methylation and/or mutations results in impaired DNA repair and often sensitivity to poly (ADP-ribose) polymerase inhibitors (PARPi)/platinum-based chemotherapies. BRCA1/2 deficiency can also modulate chromatin accessibility and enhancers of drug-resistant and/or DNA damage response (DDR)-associated genes. Reactivation of BRCA1/2 either by demethylation or reversion mutations can result in tumour cells with functional DDR. Ultimately, these mechanisms result in tumour cells that are drug resistant, have functional DDR and no longer respond to previous PARPi/platinum chemotherapy.BRCA1/2 deficiency can impair the HR pathway leading to increased sensitivity to DNA-damaging agents such as platinum chemotherapy and PARPi. However, BRCA1/2 deficiency can also lead to chromatin remodelling and reduced H3K27ac at regulatory regions such as enhancers/super-enhancers, leading to the aberrant expression of genes including those associated with drug resistance/DDR-associated genes. Reactivation of BRCA1/2 and/or other DDR-associated genes can result in tumour cells which are drug resistant and have functional DNA damage repair.
2. Can Epigenetic Therapies Reverse Epigenetically Driven Drug Resistance?
The clinically relevant examples above demonstrate how promoter DNA methylation may confer tumour chemosensitivity or chemoresistance. However, there is a wide spectrum of DDR genes whose epigenetic regulation can influence chemosensitivity [2]. Epigenetic therapies such as DNA-demethylating agents and inhibitors of maintenance of histone post-translational modification (for instance, histone deacetylase inhibitors) are now registered for clinical use, particularly in haematological malignancies. Furthermore, they remain the focus of many clinical trials in epithelial cancers [30]. However, careful patient selection will be key to demonstrating clinical efficacy of epigenetic therapies, especially when used in combination with other therapies.
This is exemplified in early clinical trials of DNA-demethylating agents in HGSOC. In cell line models, the loss of MMR due to MLH1 methylation results in failure to engage apoptotic responses and resistance to platinum coordination complexes and alkylating agents which can be reversed by DNA-demethylating agents such as 5-azacytidine and its derivatives [31]. DNA methylation has been used as a pharmacodynamic marker in surrogate tissues, such as blood, to demonstrate biological activity and guide the scheduling of combination studies with other therapies [32]. There have been two randomised phase II studies of DNA-demethylating agents and carboplatin in HGSOC with contrasting outcomes [33][34]. One study was closed early due to unacceptable toxicity and lack of efficacy of the combination compared to single-agent carboplatin [33]. The other trial showed an improvement in the 6-month progression-free survival of patients treated with the combination. However, this trial did not show statistically significant superiority for the primary endpoint of progression-free survival, potentially due to being statistically underpowered for the latter endpoint [34]. Both studies explored the combination of carboplatin with a DNA-demethylating agent during second-line chemotherapy. Glasspool et al., recruited partially platinum-sensitive patients recurring 6–12 months following the initial response to platinum-based chemotherapy while Oza et al., recruited women with recurrence within 6 months of the last platinum-containing regimen. It is possible that partially platinum sensitive patients may have a different proportion of women with tumours who are sensitive due to the methylation of HR genes, such as BRCA1, and for whom a demethylating agent may have an adverse effect. Neither study selected patient recruitment based on the methylation status of the patients’ tumours.
As previously mentioned, BRCA1 is frequently methylated in high-grade serous ovarian cancer and associated with increased patient survival following platinum-based and PARP inhibitor chemotherapy compared to patients with HR-proficient tumours. BRCA1/2 deficiency remains the strongest predictor of PARPi sensitivity [35] although abrogation of other key HR genes including FA Complementation Group A (FANCA) [36], DNA Repair Protein RAD51 homolog 1 (RAD51) [37], X-ray Repair Cross Complementing 2 (XRCC2) and X-ray Repair Cross Complementing 3 (XRCC3) [38] and DNA Polymerase Delta 4 (POLD4) [39] have been linked to platinum and/or PARPi responses. Furthermore, not all BRCA mutant tumours are HR deficient and many HR-proficient tumours can initially respond well to PARPi [40][41][42][43][44] which has been attributed to the involvement of PARP in other non-DDR associated mechanisms including chromatin remodelling [45]. Unfortunately, as with platinum-based chemotherapy, primary and acquired resistance to PARPi is common [35][40][46][47]. Reversion mutations can restore the function of HR-associated genes frequently mutated in HGSOC, including BRCA1/2 [48][49][50][51] and RAD51C/D [52]. Epigenetic mechanisms including histone modifications may also contribute to PARPi resistance although the exact mechanisms remain poorly understood [12][52][53][54][55][56][57].
Histone methylation has been linked to PARP inhibitor sensitivity in multiple cancers [58]. Enhancer of Zeste Homolog 2 (EZH2) and Euchromatic Histone Lysine Methyltransferase 2 (EHMT2) both maintain repressive H3K27 and H3K9 methylation histone marks, respectively, and are frequently overexpressed in cancer [59]. The inhibition of EZH2 alone has previously been linked to reducing the expression of multiple genes associated with DDR pathways in multiple cancers including prostate [60] and ovarian [61]. Furthermore, EZH2 inhibition has been shown to sensitise breast cancer cells to PARPi [58][61] and PARPi can regulate EZH2 expression [62] via PARPylation. EHMT2 has been linked with directly recruiting HR-associated factors, including BRCA1 to promote DNA damage repair [63]. The inhibition of EHMT2 promotes increased DNA damage and altered cell cycle regulation [64] and PARPi resistant cells treated with an EHMT1/2 inhibitor show significantly altered gene expression changes enriched in pro-survival pathways including, phosphatidylinositol 3 kinase(PI3K), protein kinase B (AKT) and mammalian target of rapamycin (mTOR) [64]. In BRCA1-depleted SUM149 breast cancer cells and PDX models, treatment with an EZH2i and PARPi reduced tumour growth more than single PARPi treatment; however, this effect was not seen in a BRCA2-depleted mouse model of breast cancer [65]. EZH2 inhibition alone may not be sufficient to modulate chromatin conformation [66] therefore dual EZH2/EHMT2 inhibitors in combination with PARPi would perhaps be of future interest. A clear theme regarding epigenetic therapies in combination with chemotherapy and their ability to modulate epigenetically driven drug resistance is that any future combination therapies must have clear stratification markers. The global epigenetic and mutational profile of DDR-associated genes must also be considered in order to maximise the positive outcomes for patients. A summary of key DDR genes associated with drug resistance and/or regulated by epigenetic mechanisms are shown in Table 1.
Table 1. Summary of DDR-associated genes, how they can be epigenetically regulated and their involvement in drug response.
| Gene | Symbol | Summary | Reference Number |
|---|---|---|---|
| O6-Methylguanine-DNA Methyltransferase | MGMT | Methylated associated with increased sensitivity to temozolomide. Enhancer region associated with increased expression and resistance to temozolomide. | [1][3][5][6][26] |
| Breast Cancer type 1 susceptibility protein | BRCA1 | Methylated associated with sensitivity to PARPi/platinum and loss of H3K27ac at enhancer regions. | [7][8][11][12][27][35] |
| Breast Cancer type 2 susceptibility protein | BRCA2 | Deficiency causes chromatin conformation changes and increased sensitivity to PARPi/platinum. | [7][8][11][12][28] |
| MutL Homolog 1 | MLH1 | Unmethylated associated with temozolomide/platinum resistance and loss of MMR. | [15][16][17][31] |
| FA Complementation Group F | FANCF | Methylation associated with sensitivity to platinum, unmethylated associated with platinum resistance. | [24][25] |
| FA Complementation Group A | FANCA | Germline mutation associated with increased sensitivity to DNA damaging agents. | [36] |
| DNA Repair Protein RAD51 homolog 1 | RAD51 | High expression associated with platinum resistance. | [37] |
| X-ray Repair Cross Complementing 2 | XRCC2 | Low expression associated with sensitivity to PARPi. | [38] |
| X-ray Repair Cross Complementing 3 | XRCC3 | Low expression associated with sensitivity to PARPi. | [38] |
| DNA Polymerase Delta 4 | POLD4 | Low expression associated with sensitivity to PARPi/platinum. | [39] |
| RAD51 Paralog C | RAD51C | Reversion mutations associated with increased resistance to PARPi. | [52] |
| RAD51 Paralog D | RAD51D | Reversion mutations associated with increased resistance to PARPi. | [52] |
| Euchromatic Histone Lysine Methyltransferase 2 | EHMT2 | Maintains repressive H3K9 methylation marks. Recruits HR-associated factors, Inhibition of EHMT2 promotes DNA damage. | [58][59][63][64][66] |
| Enhancer of Zeste Homolog 2 | EZH2 | Maintains repressive H3K27 methylation marks. Controls expression of multiple DDR-associated genes. Inhibition of EZH2 sensitises cells to PARPi. | [58][59][60][61][66] |
3. Epigenetic Changes in Normal Tissue following Chemotherapy
The observations of increased DNA methylation at gene promoters in drug resistant tumours and cell line models following chemotherapy treatment could be due to the selection of cells epigenetically silenced that are present in the tumour before chemotherapy. Alternatively, DNA damage induced by the chemotherapy may be causing methylation changes. DNA damage such as platinum-induced and DNA double-strand breaks are recognized by DNA mismatch repair proteins [67]. These bind and recruit the DNA methylating enzyme encoded by the DNMT1 gene, resulting in aberrant DNA methylation [68][69][70][71]. At the time of relapse following platinum-based chemotherapy, changes in methylation at specific CpG sites in blood DNA are observed which mirror changes occurring in tumour DNA at relapse in ovarian cancer patients [72]. These changes can predict clinical outcome and identify patients with better overall survival. In contrast, blood samples taken at presentation prior to treatment show no association between methylation and survival. DNA methylation at specific CpGs in blood has been associated with environmental exposures including smoking and alcohol consumption [73][74]. Smoking-induced methylation changes at the ARRH gene have been associated with aberrant ARRH transcription in lung epithelial cells [60] and mediate the risk of developing lung cancer [75]. In 2020, it was estimated that 4.1% of all cancers can be attributed to alcohol consumption [76]. A 144 CpG DNA methylation signature has been used to identify heavy alcohol consumption in whole blood samples [77]. Whilst the functional consequences of methylation changes in blood remain unknown, they may be acting as a surrogate markers for changes in a more relevant tissue. Similarly, epigenetic changes occurring in normal tissues due to DNA-damaging agents such as chemotherapy can have long-term consequences for secondary tumours or altering immune responses [78].
References
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797.
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6.
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715.
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro Oncol. 2019, 21, 167–168.
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; Van Den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51.
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.
- Stronach, E.A.; Paul, J.; Timms, K.M.; Hughes, E.; Brown, K.; Neff, C.; Perry, M.; Gutin, A.; El-Bahrawy, M.; Steel, J.H.; et al. Biomarker assessment of HR deficiency, tumor BRCA1/2 mutations, and CCNE1 copy number in ovarian cancer: Associations with clinical outcome following platinum monotherapy. Mol. Cancer Res. 2018, 16, 1103–1111.
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487.
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970.
- Bonadio, R.; Fogace, R.; Miranda, V.; Diz, M. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics 2018, 73, e450s.
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.S.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 2014, 14, 747–753.
- Wu, F.; Lu, M.; Qu, L.; Li, D.Q.; Hu, C.H. DNA methylation of hMLH1 correlates with the clinical response to cisplatin after a surgical resection in Non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 5457–5463.
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.M.; Rizzo, S.; Van Der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576.
- Gallitto, M.; Cheng He, R.; Inocencio, J.F.; Wang, H.; Zhang, Y.; Deikus, G.; Wasserman, I.; Strahl, M.; Smith, M.; Sebra, R.; et al. Epigenetic preconditioning with decitabine sensitizes glioblastoma to temozolomide via induction of MLH1. J. Neurooncol. 2020, 147, 557–566.
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis 2006, 27, 682–692.
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487.
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16.
- Marsit, C.J.; Liu, M.; Nelson, H.H.; Posner, M.; Suzuki, M.; Kelsey, K.T. Inactivation of the Fanconi anemia/BRCA pathway in lung and oral cancers: Implications for treatment and survival. Oncogene 2004, 23, 1000–1004.
- Narayan, G.; Arias-Pulido, H.; Nandula, S.V.; Basso, K.; Sugirtharaj, D.D.; Vargas, H.; Mansukhani, M.; Villella, J.; Meyer, L.; Schneider, A.; et al. Promoter Hypermethylation of FANCF: Disruption of Fanconi Anemia-BRCA Pathway in Cervical Cancer. Cancer Res. 2004, 64, 2994–2997.
- Lim, S.L.; Smith, P.; Syed, N.; Coens, C.; Wong, H.; Van Der Burg, M.; Szlosarek, P.; Crook, T.; Green, J.A. Promoter hypermethylation of FANCF and outcome in advanced ovarian cancer. Br. J. Cancer 2008, 98, 1452–1456.
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574.
- D’Andrea, A.D. The Fanconi Anemia/BRCA Signaling Pathway: Disruption in Cisplatin-Sensitive Ovarian Cancers. Cell Cycle 2003, 2, 289–291.
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.-H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949.
- Zhang, X.; Wang, Y.; Chiang, H.-C.; Hsieh, Y.-P.; Lu, C.; Park, B.H.; Jatoi, I.; Jin, V.X.; Hu, Y.; Li, R. BRCA1 mutations attenuate super-enhancer function and chromatin looping in haploinsufficient human breast epithelial cells. Breast Cancer Res. 2019, 21, 51.
- Gruber, J.J.; Chen, J.; Geller, B.; Jäger, N.; Lipchik, A.M.; Wang, G.; Kurian, A.W.; Ford, J.M.; Snyder, M.P. Chromatin Remodeling in Response to BRCA2-Crisis. Cell Rep. 2019, 28, 2182–2193.e6.
- Hung, S.; Saiakhova, A.; Faber, Z.J.; Bartels, C.F.; Neu, D.; Bayles, I.; Ojo, E.; Hong, E.S.; Pontius, W.D.; Morton, A.R.; et al. Mismatch repair-signature mutations activate gene enhancers across human colorectal cancer epigenomes. eLife 2019, 8, e40760.
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176.
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044.
- Appleton, K.; Mackay, H.J.; Judson, I.; Plumb, J.A.; McCormick, C.; Strathdee, G.; Lee, C.; Barrett, S.; Reade, S.; Jadayel, D.; et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J. Clin. Oncol. 2007, 25, 4603–4609.
- Glasspool, R.M.; Brown, R.; Gore, M.E.; Rustin, G.J.S.; McNeish, I.A.; Wilson, R.H.; Pledge, S.; Paul, J.; MacKean, M.; Hall, G.D.; et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer 2014, 110, 1923–1929.
- Oza, A.M.; Matulonis, U.A.; Alvarez Secord, A.; Nemunaitis, J.; Roman, L.D.; Blagden, S.P.; Banerjee, S.; McGuire, W.P.; Ghamande, S.; Birrer, M.J.; et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin. Cancer Res. 2020, 26, 1009–1016.
- Bouwman, P.; Jonkers, J. Molecular pathways: How can BRCA-mutated tumors become resistant to PARP inhibitors? Clin. Cancer Res. 2014, 20, 540–547.
- Wilkes, D.C.; Sailer, V.; Xue, H.; Cheng, H.; Collins, C.C.; Gleave, M.; Wang, Y.; Demichelis, F.; Beltran, H.; Rubin, M.A.; et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Mol. Case Stud. 2017, 3, a001487.
- Hoppe, M.M.; Jaynes, P.; Wardyn, J.D.; Upadhyayula, S.S.; Tan, T.Z.; Lie, S.; Lim, D.G.Z.; Pang, B.N.K.; Lim, S.P.S.; Yeong, J.; et al. Quantitative imaging of RAD51 expression as a marker of platinum resistance in ovarian cancer. EMBO Mol. Med. 2021, 13, e13366.
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599.
- Zhang, S.; Chao, H.H.; Wang, X.; Zhang, Z.; Lee, E.Y.C.; Lee, M.Y.W.T. Loss of the p12 subunit of DNA polymerase delta leads to a defect in HR and sensitization to PARP inhibitors. DNA Repair 2019, 73, 64–70.
- Ledermann, J.A.; El-Khouly, F. PARP inhibitors in ovarian cancer: Clinical evidence for informed treatment decisions. Br. J. Cancer 2015, 113, S10–S16.
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203.
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019.
- Kim, D.S.; Camacho, C.V.; Nagari, A.; Malladi, V.S.; Challa, S.; Kraus, W.L. Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol. Cell 2019, 75, 1270–1285.e14.
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940.
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562.
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251.
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart, M. An update on PARP inhibitors—Moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41.
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386.
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669.
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015.
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219.
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017, 7, 984–998.
- Curry, E.; Zeller, C.; Masrour, N.; Patten, D.K.; Gallon, J.; Wilhelm-Benartzi, C.S.; Ghaem-Maghami, S.; Bowtell, D.D.; Brown, R. Genes predisposed to DNA hypermethylation during acquired resistance to chemotherapy are identified in ovarian tumors by bivalent chromatin domains at initial diagnosis. Cancer Res. 2018, 78, 1383–1391.
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081.
- Sun, J.; Cai, X.; Yung, M.M.; Zhou, W.; Li, J.; Zhang, Y.; Li, Z.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S.; et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene 2019, 38, 564–580.
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104.
- Andronikou, C.; Rottenberg, S. Studying PAR-Dependent Chromatin Remodeling to Tackle PARPi Resistance. Trends Mol. Med. 2021, 27, 630–642.
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.S.; Hsu, J.L.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018, 37, 208–217.
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-β 1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583.
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013, 4, 331–341.
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6.
- Caruso, L.B.; Martin, K.A.; Lauretti, E.; Hulse, M.; Siciliano, M.; Lupey-Green, L.N.; Abraham, A.; Skorski, T.; Tempera, I. Poly(ADP-ribose) Polymerase 1, PARP1, modifies EZH2 and inhibits EZH2 histone methyltransferase activity after DNA damage. Oncotarget 2018, 9, 10585–10605.
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063.
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenet. 2019, 11, 165.
- Chen, M. Efficacy of PARP inhibition combined with EZH2 inhibition depends on BRCA mutation status and microenvironment in breast cancer. FEBS J. 2021, 288, 2884–2887.
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenet. 2015, 7, 84.
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98.
- O’Hagan, H.M.; Wang, W.; Sen, S.; DeStefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619.
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 2008, 4, e1000155.
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O’Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254.
- Cuozzo, C.; Porcellini, A.; Angrisano, T.; Morano, A.; Lee, B.; Di Pardo, A.; Messina, S.; Iuliano, R.; Fusco, A.; Santillo, M.R.; et al. DNA Damage, Homology-Directed Repair, and DNA Methylation. PLoS Genet. 2007, 3, e110.
- Flanagan, J.M.; Wilson, A.; Koo, C.; Masrour, N.; Gallon, J.; Loomis, E.; Flower, K.; Wilhelm-Benartzi, C.; Hergovich, A.; Cunnea, P.; et al. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in patients with ovarian cancer. Clin. Cancer Res. 2017, 23, 2213–2222.
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation Into Cancer And Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851.
- Flanagan, J.M.; Brook, M.N.; Orr, N.; Tomczyk, K.; Coulson, P.; Fletcher, O.; Jones, M.E.; Schoemaker, M.J.; Ashworth, A.; Swerdlow, A.; et al. Temporal stability and determinants of white blood cell DNA methylation in the breakthrough generations study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 221–229.
- Fasanelli, F.; Baglietto, L.; Ponzi, E.; Guida, F.; Campanella, G.; Johansson, M.; Grankvist, K.; Johansson, M.; Assumma, M.B.; Naccarati, A.; et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat. Commun. 2015, 6, 10192.
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.E.P.P.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 2021, 22, 1071–1080.
- Liu, C.; Marioni, R.E.; Hedman, A.K.; Pfeiffer, L.; Tsai, P.C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.; Tanaka, T.; et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433.
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
850
Revisions:
2 times
(View History)
Update Date:
28 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No