 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio Musarò | + 2072 word(s) | 2072 | 2022-03-17 03:22:39 | | | |
| 2 | Jessie Wu | + 3 word(s) | 2075 | 2022-03-28 03:01:57 | | | | |
| 3 | Jessie Wu | + 3 word(s) | 2075 | 2022-03-28 03:03:22 | | | | |
| 4 | Jessie Wu | Meta information modification | 2075 | 2022-03-31 07:59:32 | | | | |
| 5 | Jessie Wu | Meta information modification | 2075 | 2022-03-31 08:07:17 | | |
Video Upload Options
One of the crucial systems severely affected in several neuromuscular diseases is the loss of effective connection between muscle and nerve, leading to a pathological non-communication between the two tissues. The neuromuscular junction (NMJ) represents the critical region at the level of which muscle and nerve communicate. Defects in signal transmission between terminal nerve endings and muscle membrane is a common feature of several physio-pathologic conditions including aging and Amyotrophic Lateral Sclerosis (ALS). Nevertheless, controversy exists on whether pathological events beginning at the NMJ precede or follow loss of motor units.
1. Introduction
Muscle and nerve communicate at the level of a specialized region, namely the neuromuscular junction (NMJ), a synaptic connection where the peripheral nervous system contacts skeletal muscle fibers, governing crucial vital processes, such as body voluntary movements and breathing [1]. Nerve activity guarantees not only muscle contraction but can induce myoblast orientation [2] and strictly influences fiber type specification and myosin isoforms expression [3]. Skeletal muscle fibers can be generally classified as fast or slow twitch, based on their contractile and metabolic properties [4].
2. Amyotrophic Lateral Sclerosis and Aging as Paradigmatic Examples of Altered Nerve-Muscle Communication
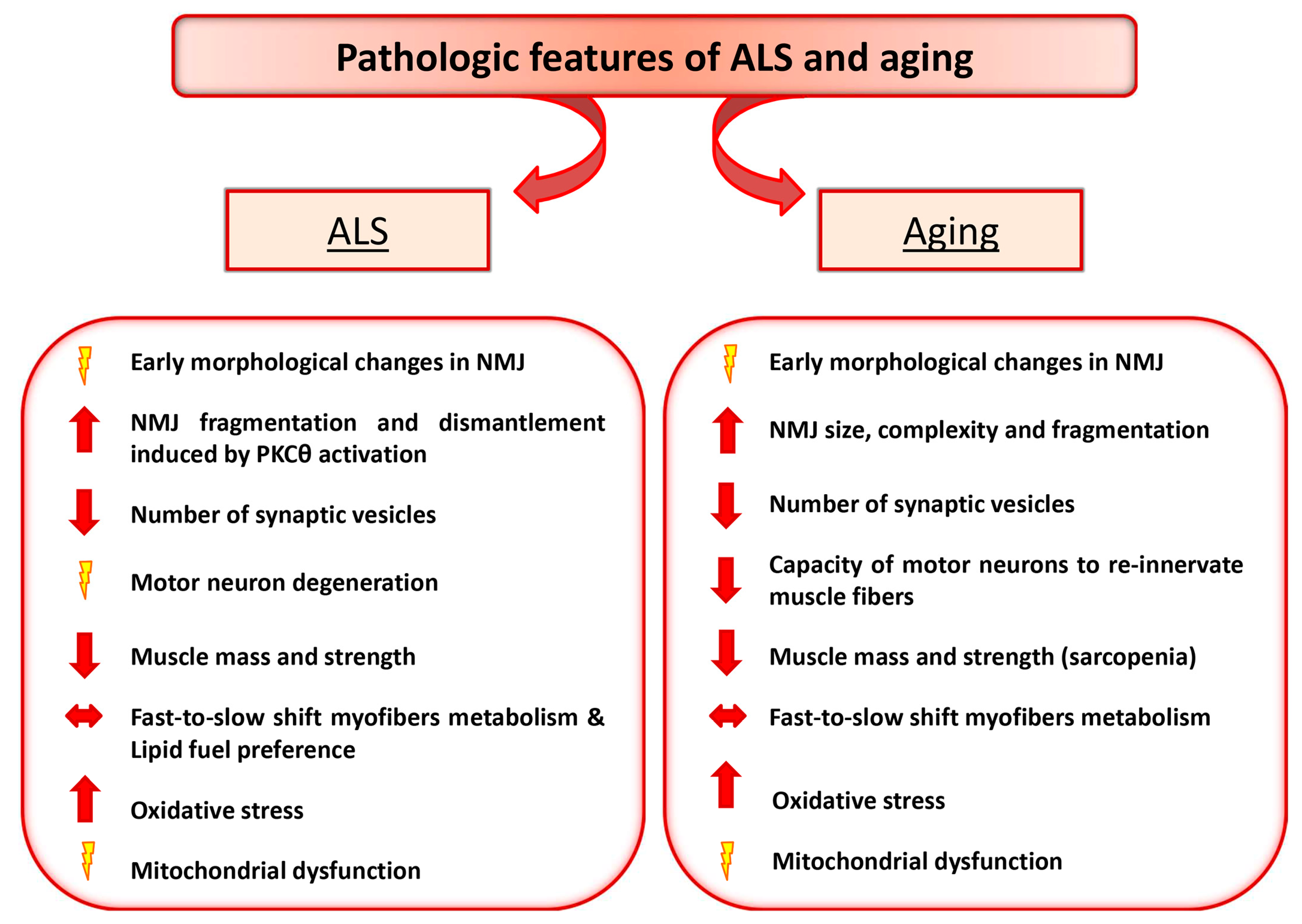
3. Motor Neuron Diseases: ALS
3.1. PKCθ as a Potential Signaling Involved in NMJ Dysfunction in ALS Disease
3.2. Metabolic Changes and Mitochondrial Alteration May Influence NMJ Stability
References
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387.
- Mège, R.M.; Goudou, D.; Giaume, C.; Nicolet, M.; Rieger, F. Is intercellular communication via gap junctions required for myoblast fusion? Cell Adhes. Commun. 1994, 2, 329–343.
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423.
- Hughes, S.M. Muscle development: Electrical control of gene expression. Curr. Biol. 1998, 8, R892–R894.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013.
- Musarò, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322.
- Park, K.H.J. Mechanisms of Muscle Denervation in Aging: Insights from a Mouse Model of Amyotrophic Lateral Sclerosis. Aging Dis. 2015, 6, 380–389.
- García, M.L.; Fernández, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26.
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44.
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal. 2018, 28, 1105–1119.
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014.
- Hegedus, J.; Putman, C.T.; Gordon, T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 154–164.
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential motor unit loss in the SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008, 586, 3337–3351.
- De Winter, F.; Vo, T.; Stam, F.J.; Wisman, L.A.B.; Bär, P.R.; Niclou, S.P.; van Muiswinkel, F.L.; Verhaagen, J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 2006, 32, 102–117.
- Chance, P.F.; Rabin, B.A.; Ryan, S.G.; Ding, Y.; Scavina, M.; Crain, B.; Griffin, J.W.; Cornblath, D.R. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am. J. Hum. Genet. 1998, 62, 633–640.
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165.
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001.
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456.
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004.
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009.
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009.
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102.
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010.
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 2019, 12.
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 2002, 22, 4825–4832.
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003.
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436.
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302.
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112.
- Rudolf, R.; Deschenes, M.R.; Sandri, M. Neuromuscular junction degeneration in muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 177–181.
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004.
- Schaefer, A.M.; Sanes, J.R.; Lichtman, J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2005, 490, 209–219.
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001.
- Horwitz, G.; Jordan, H.; Krekelberg, B.; Richert, M.; Reyes, J.; Foundation, T.S.; Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417.
- Martinez-Pena, Y.; Valenzuela, I.; Pires-Oliveira, M.; Akaaboune, M. PKC and PKA regulate AChR dynamics at the neuromuscular junction of living mice. PLoS ONE 2013.
- Li, M.-X.; Jia, M.; Yang, L.-X.; Jiang, H.; Lanuza, M.A.; Gonzalez, C.M.; Nelson, P.G. The role of the theta isoform of protein kinase C (PKC) in activity-dependent synapse elimination: Evidence from the PKC theta knock-out mouse in vivo and in vitro. J. Neurosci. 2004, 24, 3762–3769.
- Camerino, G.M.; Bouchè, M.; De Bellis, M.; Cannone, M.; Liantonio, A.; Musaraj, K.; Romano, R.; Smeriglio, P.; Madaro, L.; Giustino, A.; et al. Protein kinase C theta (PKCθ) modulates the ClC-1 chloride channel activity and skeletal muscle phenotype: A biophysical and gene expression study in mouse models lacking the PKCθ. Pflügers Arch. Eur. J. Physiol. 2014, 466, 2215–2228.
- Serra, C.; Federici, M.; Buongiorno, A.; Senni, M.I.; Morelli, S.; Segratella, E.; Pascuccio, M.; Tiveron, C.; Mattei, E.; Tatangelo, L.; et al. Transgenic mice with dominant negative PKC-theta in skeletal muscle: A new model of insulin resistance and obesity. J. Cell. Physiol. 2003, 196, 89–97.
- Kasarskis, E.J.; Neville, H.E. Management of ALS: Nutritional care. Neurology 1996, 47, S118–S120.
- Desport, J.-C.; Torny, F.; Lacoste, M.; Preux, P.-M.; Couratier, P. Hypermetabolism in ALS: Correlations with Clinical and Paraclinical Parameters. Neurodegener. Dis. 2005, 2, 202–207.
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117.
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242.
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.-L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009.
- Huisman, M.H.B.; Seelen, M.; van Doormaal, P.T.C.; de Jong, S.W.; de Vries, J.H.M.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Effect of Presymptomatic Body Mass Index and Consumption of Fat and Alcohol on Amyotrophic Lateral Sclerosis. JAMA Neurol. 2015, 72, 1155–1162.
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener. Dis. 2004, 1, 245–254.
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.-L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.-P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546.
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic Changes Associated with Muscle Expression of SOD1G93A. Front. Physiol. 2018, 9, 831.
- Carrì, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599.
- Shi, P.; Wei, Y.; Zhang, J.; Gal, J.; Zhu, H. Mitochondrial Dysfunction is a Converging Point of Multiple Pathological Pathways in Amyotrophic Lateral Sclerosis. J. Alzheimer’s Dis. 2010, 20, S311–S324.
- Barrett, E.F.; Barrett, J.N.; David, G. Dysfunctional mitochondrial Ca2+ handling in mutant SOD1 mouse models of fALS: Integration of findings from motor neuron somata and motor terminals. Front. Cell. Neurosci. 2014, 8, 184.
- Villegas, R.; Martinez, N.W.; Lillo, J.; Pihan, P.; Hernandez, D.; Twiss, J.L.; Court, F.A. Calcium Release from Intra-Axonal Endoplasmic Reticulum Leads to Axon Degeneration through Mitochondrial Dysfunction. J. Neurosci. 2014, 34, 7179–7189.
- Talbot, J.D.; Barrett, J.N.; Nonner, D.; Zhang, Z.; Wicomb, K.; Barrett, E.F. Preservation of neuromuscular function in symptomatic SOD1-G93A mice by peripheral infusion of methylene blue. Exp. Neurol. 2016, 285, 96–107.
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390.
- Mohajeri, M.H.; Figlewicz, D.A.; Bohn, M.C. Intramuscular Grafts of Myoblasts Genetically Modified to Secrete Glial Cell Line-Derived Neurotrophic Factor Prevent Motoneuron Loss and Disease Progression in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Hum. Gene Ther. 1999, 10, 1853–1866.
- Acsadi, G.; Anguelov, R.A.; Yang, H.; Toth, G.; Thomas, R.; Jani, A.; Wang, Y.; Ianakova, E.; Mohammad, S.; Lewis, R.A.; et al. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum. Gene Ther. 2002, 13, 1047–1059.
- Kaspar, B.K. Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science 2003, 301, 839–842.
- Rabinovsky, E.D.; Gelir, E.; Gelir, S.; Lui, H.; Kattash, M.; Demayo, F.J.; Shenaq, S.M.; Schwartz, R.J. Targeted expression of IGF-1 transgene to skeletal muscle accelerates muscle and motor neuron regeneration. FASEB J. 2003, 17, 53–55.
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199.
- Ozdinler, P.H.; Macklis, J.D. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 2006, 9, 1371–1381.
- Li, S.; Li, Y.; Yu, S.; Du, W.; Zhang, L.; Dai, Y.; Liu, Y.; Li, N. Expression of insulin-like growth factors systems in cloned cattle dead within hours after birth. Mol. Reprod. Dev. 2007, 74, 397–402.
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; L Sweeney, H.; Rosenthal, N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200.
- Sakowski, S.A.; Schuyler, A.D.; Feldman, E.L. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 63–73.
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: Results of a randomized, double-blind, controlled study. Sci. Rep. 2019, 9, 2837.
- Witzemann, V.; Brenner, H.R.; Sakmann, B. Neural factors regulate AChR subunit mRNAs at rat neuromuscular synapses. J. Cell Biol. 1991, 114, 125–141.
- Sengupta-Ghosh, A.; Dominguez, S.L.; Xie, L.; Barck, K.H.; Jiang, Z.; Earr, T.; Imperio, J.; Phu, L.; Budayeva, H.G.; Kirkpatrick, D.S.; et al. Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis. 2019, 124, 340–352.
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786.
- Arnold, A.-S.; Gill, J.; Christe, M.; Ruiz, R.; McGuirk, S.; St-Pierre, J.; Tabares, L.; Handschin, C. Morphological and functional remodelling of the neuromuscular junction by skeletal muscle PGC-1α. Nat. Commun. 2014, 5, 3569.

