One of the crucial systems severely affected in several neuromuscular diseases is the loss of effective connection between muscle and nerve, leading to a pathological non-communication between the two tissues. The neuromuscular junction (NMJ) represents the critical region at the level of which muscle and nerve communicate. Defects in signal transmission between terminal nerve endings and muscle membrane is a common feature of several physio-pathologic conditions including aging and Amyotrophic Lateral Sclerosis (ALS). Nevertheless, controversy exists on whether pathological events beginning at the NMJ precede or follow loss of motor units.
1. Introduction
Muscle and nerve communicate at the level of a specialized region, namely the neuromuscular junction (NMJ), a synaptic connection where the peripheral nervous system contacts skeletal muscle fibers, governing crucial vital processes, such as body voluntary movements and breathing [1]. Nerve activity guarantees not only muscle contraction but can induce myoblast orientation [2] and strictly influences fiber type specification and myosin isoforms expression [3]. Skeletal muscle fibers can be generally classified as fast or slow twitch, based on their contractile and metabolic properties [4].
2. Amyotrophic Lateral Sclerosis and Aging as Paradigmatic Examples of Altered Nerve-Muscle Communication
The impaired neuromuscular transmission represents a critical feature of several pathological conditions in which structural changes in NMJ might contribute to muscle weakness, altered motor neuron activity, and loss of muscle fibers.
Aging represents a physiologic and progressive decay of the homeostatic processes of the entire organism and it is broadly defined as the time-dependent functional decline that affects most living organisms. Common denominators of aging in different organisms include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [
18].
ALS is a complex and severe disease associated with numerous pathologic mechanisms, including oxidative stress, mitochondrial dysfunction, axonal damage, microglial activation, inflammation, excitotoxicity, and protein aggregation [
19].
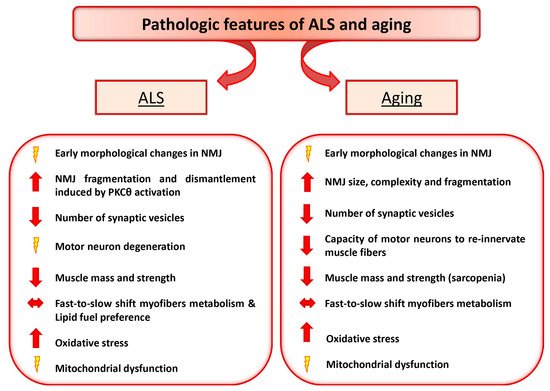
Interestingly, although aging and ALS display different progressive loss of physiological integrity, they share some common pathologic feature, including high levels of oxidative damage, decreased number of synaptic vesicles, reduced and altered mitochondria in the plaque region [
20,
21,
22,
23,
24] (
Figure 1). Moreover, an early pathologic sign observed in both ALS and aging is the morphologic alteration of NMJ. However, whether changes in the NMJ precede or follow loss of motor units remains unresolved. Here we detail the NMJ degenerative features in ALS and aging and discuss the paradigm that retrograde signaling, from muscle to nerve, might represent an early event preceding motor neuron degeneration.
Figure 1. Amyotrophic Lateral Sclerosis (ALS) and Aging share some common pathologic features. The diagram depicts the relevant pathologic changes observed in ALS and aging.
3. Motor Neuron Diseases: ALS
One of the best examples of impaired interplay between nerve and muscle is ALS, a fatal disease characterized by motor neurons degeneration, muscle atrophy, weakness and, ultimately, muscle paralysis with respiratory failure. During ALS progression muscle denervation is accompanied by changes in muscle fiber profile with a preference loss of fast-twitch fiber [
25,
26], due to higher vulnerability of fast-fatigable innervating motor neurons and to specific signals released by PSCs [
27]. ALS is epidemiologically classified into two forms: sporadic (90–95%) and familial (5–10%) form. Among the familial cases, approximately 20% correlate with mutations in the sequence of the Superoxide Dismutase 1 gene (
SOD1), which encodes for an important antioxidant enzyme. In addition to
SOD1 mutations, other gene defects have been reported to cause ALS, including
senataxin (
SETX) [
28],
alsin [
29,
30],
dynactin [
31],
synaptobrevin/VAMP (vesicle-associated membrane protein)-associated protein B (
VAPB) [
32],
TDP-43, FUS/TLS [
33,
34],
profilin (
PFN1) [
35],
MATR3,
CHCHD10,
TBK1,
TUBA4A,
NEK1,
C21orf2, and
CCNF [
36]. However, even in these cases, where a well-defined mutation has been linked to the disease, a clear correlation between the genetic defect and the pathophysiology of the disease has not yet been disclosed.
Much of what we presently know about the role of mutant genes in ALS is based on studies of transgenic animals in which the potential genes involved in ALS are overexpressed under the control of specific promoters. Nevertheless, the failure to translate the positive results obtained in animal models into successful trials in human has cooled the enthusiasms and raised important questions on the validity of either animal models or methodological approaches. Thus, robust criteria and guidelines for preclinical animal research in ALS are necessary [
37]. One of the experimental models that has been widely used in ALS-related studies is the transgenic mutant SOD1 mouse. Although the SOD1 mutant mice present some limitations compared to ALS patients it remains, along with other ALS-related mice, an ideal model for preclinical tests and proof-of-concept studies [
38]. The obvious loss of motor neurons in the spinal cord initially focused attention on how mutant SOD1 may act within motor neurons to provoke neuronal degeneration and death. Among pathogenic events, glutamate-induced excitotoxicity, oxidative stress, protein aggregation, and mitochondrial dysfunction within motor neurons have been proposed. However, the mutant gene products are widely expressed, raising the possibility that the toxic cascade may be achieved wholly or in part by mutant SOD1 action in non-neuronal cells. Notably, restriction of
SOD1 mutant expression selectively to post-natal motor neurons failed to produce detectable sign of pathology or motor-neuron disease [
39], suggesting that other cell types may be involved in ALS-associated neurodegeneration. Indeed, analysis of chimeras generated between wild type and SOD1 mutant mouse embryonic cells revealed that wild type non neuronal cells in adult chimeric animals extended the survival of SOD1 mutant motor neurons [
40].
The generation of mouse models expressing human mutated
SOD1 gene (
SOD1G93A), exclusively in skeletal muscle, added new insights into the potential primary targets of the mutant SOD1 toxic protein. Muscle-specific expression of mutant
SOD1G93A caused accumulation of reactive oxygen species (ROS), mitochondria dysfunction, muscle atrophy, NMJ dismantlement, microgliosis [
23,
41], and neuron degeneration [
42]. All together this evidence suggests that local toxic effect of
SOD1G93A is a primary determinant of ALS-associated muscle pathology and that retrograde signals from muscle to nerve may contribute, in a sort of dying back phenomenon, to synapse and axon damage [
43,
44]. In fact, several other recent evidences confirmed the hypothesis that motor neurons are not the only primary targets of SOD1
G93A-mediated toxicity and revealed that early changes of neuromuscular transmission start long before motor symptoms onset. Fischer and colleagues demonstrated that pathological changes of motor neuron disease begin at the distal axon and they observed denervation and reinnervation changes in muscle tissue without any pathological signs in neurons cells [
45]. Furthermore, Schafer’s group characterized two different types of motor neurons, the “losers” or denervated branches, and the “compensators”, or reinnervating branches, which display different “susceptibility” to the toxic properties of
SOD1G93A mutant gene product [
46]. Although a conclusive link is still missing, it is intriguing to speculate that loser and compensator neurons are subject to different influences from neighboring interneurons, astrocytes, or microglia [
40] and from the vasculature [
47,
48] and muscle fibers they innervate, all of which might provide either toxic or protective factors.
This evidence supports the notion that NMJ dismantlement can occur independently from motor neuron degeneration and may represent an early pathogenic signature of muscle-nerve communication defects.
3.1. PKCθ as a Potential Signaling Involved in NMJ Dysfunction in ALS Disease
It has been demonstrated that Protein Kinase C and A (PKC and PKA) activities have antagonistic effects on NMJ stability and Acetylcholine Receptors (AChRs) recycling [
49]. Stimulation of PKC or inactivation of PKA significantly accelerates the removal of postsynaptic AChRs and depresses AChR recycling. A recent work disclosed the PKC isoform, namely PKCθ, which triggers NMJ dismantlement in a mouse model expressing
SOD1G93A mutant gene. PKCθ is a kinase that triggers in physiological condition the dismantlement of supernumerary NMJs during the first postnatal days [
23,
50]. It has been demonstrated that perturbation in redox signaling cascades, induced by muscle-specific accumulation of mutant
SOD1G93A gene, promotes the activation of PKCθ that in turn acts as mediator of endplates destabilization [
23]. In contrast, PKCθ selective pharmacological inhibition reduces oxidative damage and guarantees the stabilization of AChR turnover and the rescue of the NMJ morphological complexity [
23].
PKCθ also modulates the activation of two sensors of nerve activity, Calcineurin (CN) and Nuclear Factor of Activated T cells (NFATc1), which promote slow muscle phenotype through the activation of their target Myocyte Enhancer Factor 2D (MEF2D), involved in muscle glucose homeostasis. Furthermore, it has been demonstrated that PKCθ-lacking mice display an impaired glucose homeostasis in fast muscles [
51,
52], suggesting that PKCθ can participate to muscle metabolic changes associated to muscle denervation and ALS disease progression.
3.2. Metabolic Changes and Mitochondrial Alteration May Influence NMJ Stability
Metabolic homeostasis and energy balance result severely compromised in both ALS patients and mouse models, where the energy expenditure is higher than the intake and it is associated to an increased energy demand and to an abnormal lipid metabolism [
53,
54,
55,
56,
57]. In fact, a recent study revealed that ALS patients start losing weight almost 10 years before the clinical onset of the disease and show a higher daily energy intake to compensate the elevated energy consumption [
58]. Likewise, the ALS mouse model that ubiquitously overexpress
SOD1G93A mutant gene shows similar energetic alterations, such as an increased energy expenditure and a concomitant skeletal muscle hypermetabolism [
59].
Further evidence has also pointed out that metabolic alterations in SOD1
G93A mouse model can be distinct from denervation [
60] and that muscle specific accumulation of SOD1
G93A is able to induce metabolic changes in glucose and lipid pathways, independently from motor neuron degeneration and preceding muscle denervation [
61]. These data suggest that some of the metabolic changes represent part of the earliest signs of morphological and functional alterations occurring in the motor nerve terminals. Among cell components, dysfunctional mitochondria represent key regulators of metabolic changes [
62,
63,
64,
65]. Indeed, treatment with mitochondria protective drug can preserve NMJ function and structure in ALS mouse model at late stages of the disease, suggesting a central role of mitochondria in the pathology [
66]. The link between muscle functional mitochondrial alterations and neurodegeneration has been clarified by Dupuis and collaborators. The analysis of a transgenic mouse model overexpressing the potent mitochondrial uncoupler protein (
UCP1) exclusively in skeletal muscle revealed that muscle specific expression of
UCP1 induces an age-dependent deterioration of the NMJ [
67]. This defect correlated with progressive signs of denervation, supporting the idea of the crucial role of skeletal muscle on nerve homeostasis and revealing the potential molecular signature associated with the dying back phenomenon [
67].
It has been demonstrated that antioxidant treatment of mice expressing human mutated
SOD1 gene (
SOD1G93A) exclusively in skeletal muscle was able to rescue mitochondrial functionality and NMJ stability [
23,
41]. Moreover, studies on SOD1
G93A mice overexpressing the neurotrophic factors GDNF or IGF-1 in muscle tissue revealed an hyperinnervation of muscle fibers [
68,
69,
70,
71,
72,
73,
74,
75,
76], NMJ stability, and motor neuron preservation [
40,
44,
47]. This suggests that a sort of “saving back” approach can be envisaged. Nevertheless, not all treatments guarantee the same efficacy. Neuromuscular Magnetic Stimulation (NMMS) was able to induce a significant increase of muscle strength, without significant change in Compound Muscle Action Potential (CMAP) amplitude, suggesting that the improvement was not related to reinnervation phenomena [
77]. In particular, NMMS activated a molecular mini-circuit to counteract muscle atrophy, to promote muscle robustness and maintenance of fiber type composition, and to induce NMJ stabilization. In addition, NMMS induced an improvement of AChR functionality, characterized by the reduction of the expression level of gamma subunit of AChR [
77], whose expression increases in denervated muscle or under conditions that alter NMJ functionality [
23,
78].
It has been also demonstrated that Musk activation preserves synapses at the NMJ in diaphragm muscles of mutant SOD1 transgenic mice, without any positive effects on motor neuron death and survival [
79]. Cleveland and colleagues demonstrated that the over expression of Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha in the skeletal muscle of ALS transgenic mice is able to increase mitochondria energy production, ameliorating muscle endurance and performance and reducing muscle atrophy, without any effects on muscle innervation and motor neuron survival [
80]. Nevertheless, the authors did not detail NMJ morphology in PGC1α overexpressing ALS mice even if it has been demonstrated that muscle-specific
PGC-1α expression induces functional improvement in NMJ [
81]. These data suggest that additional work is necessary to define the best experimental conditions to support a “saving back” therapeutic approach. Moreover, all these data reinforce the evidence that ALS is a complex multi-systemic disease in which alterations in structural, physiological and metabolic parameters in different cell types, including motor neurons, glia, NMJ, vasculature, and muscle, act synergistically to exacerbate the disease. Moreover, all these works suggest that a single treatment or the modulation of a single pathway may be not enough to significantly counteract motor neuron degeneration and increase survival. Combinatorial approaches on different tissue targets might be necessary to achieve satisfactory therapeutic benefits.
This entry is adapted from the peer-reviewed paper 10.3390/cells8080906