 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Angela Patti | + 3882 word(s) | 3882 | 2022-03-08 08:43:56 | | | |
| 2 | Bruce Ren | -2 word(s) | 3880 | 2022-03-25 03:51:13 | | |
Video Upload Options
The ability of lipases to display activity beyond their physiological reactions, so-called “catalytic promiscuity”, has gained increasing interest in the last two decades as an important tool for expanding the application of these enzymes in organic synthesis. Some lipases have been shown to be effective in catalyzing a variety of C-C bond formation reactions and most of the investigations have been directed to the optimization of the products yield through a careful tuning of the experimental parameters. Despite the fact that new stereogenic carbons are formed in many of the tested reactions, the target products have been often obtained in racemic form and examples of an efficient asymmetric induction by the used lipases are quite limited.
1. Introduction
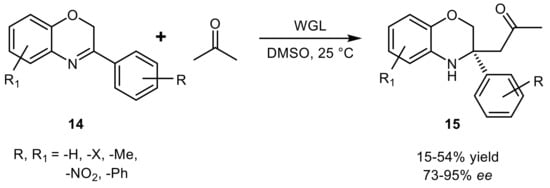
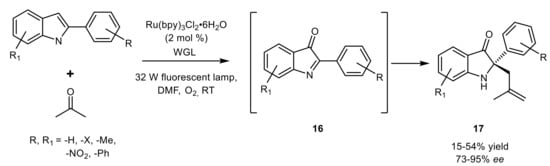
2. Lipase-Catalyzed Aldol Reactions
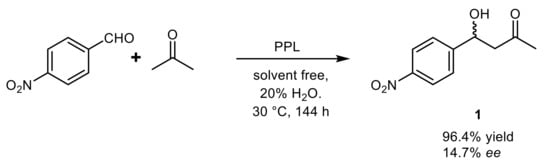
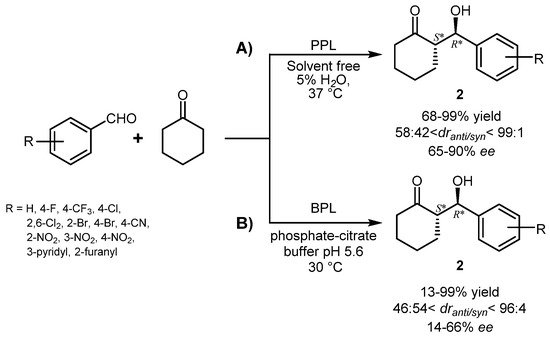
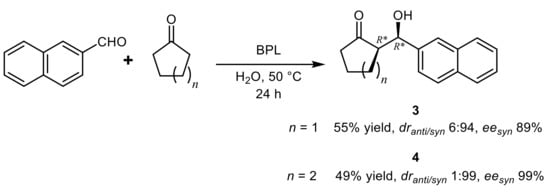
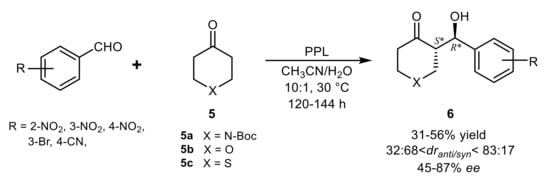
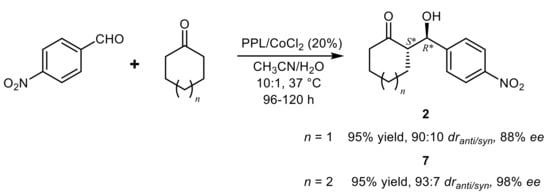
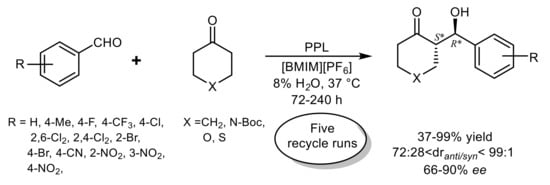
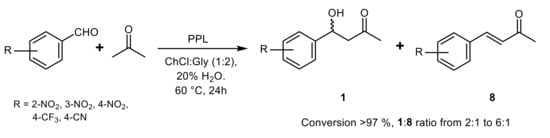
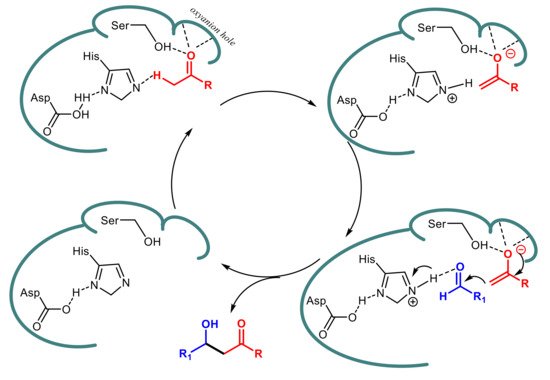
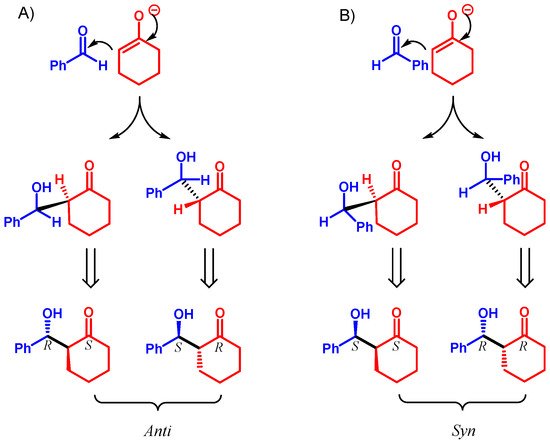
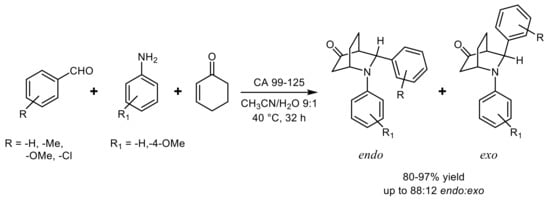
3. Lipase-Catalyzed Michael Reactions
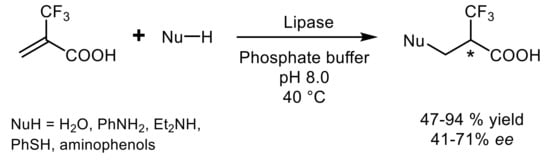
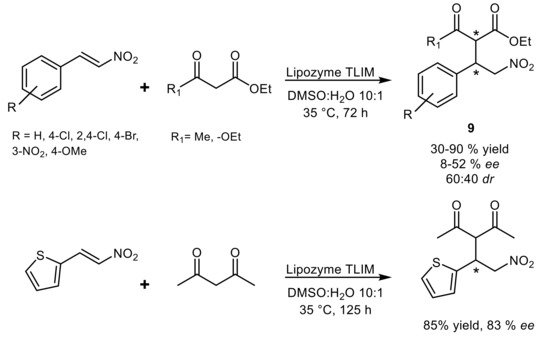
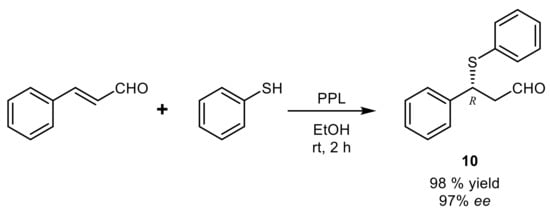
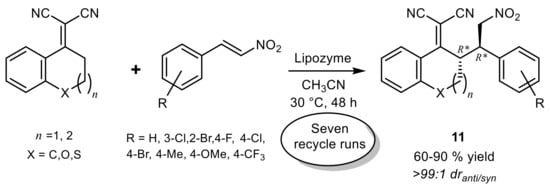
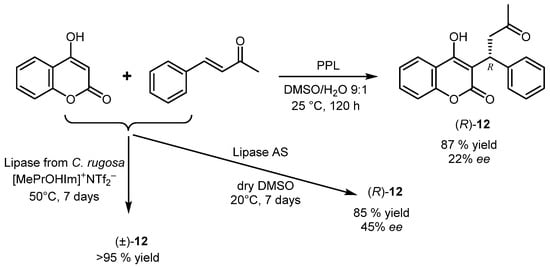
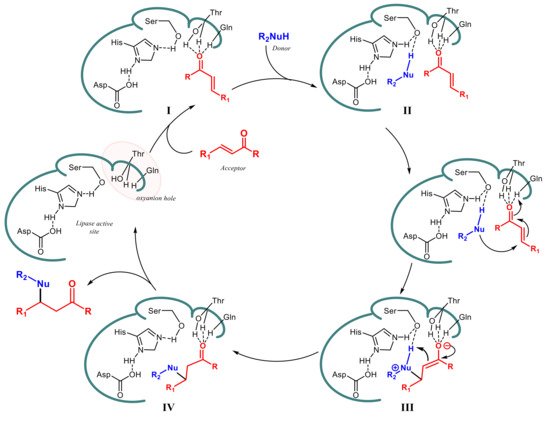
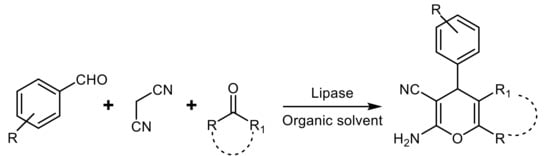
4. Lipase-Catalyzed Multicomponent Reactions
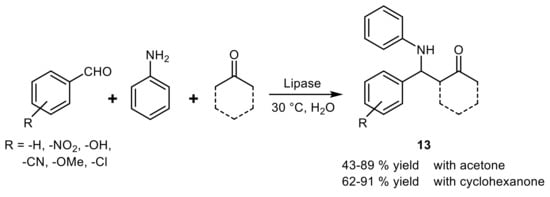
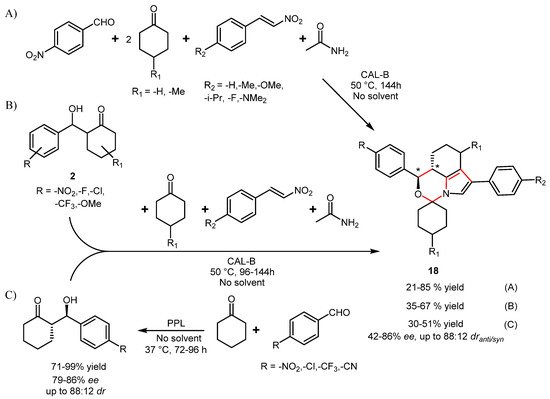
References
- Tao, J.A.; Kazlauskas, R.J. Biocatalysis for Green Chemistry and Chemical Process Development, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011.
- Sheldon, R.A.; Brady, D. Broadening the scope of biocatalysis in sustainable organic synthesis. ChemSusChem 2019, 12, 2859–2881.
- Turner, N.J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573.
- Muñoz Solano, D.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207.
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119.
- Hult, K.; Berglund, P. Enzyme promiscuity: Mechanism and applications. Trends in Biotechnol. 2007, 25, 231–238.
- Busto, E.; Gotor-Fernandez, V.; Gotor, V. Hydrolases: Catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, 39, 4504–4523.
- Humble, M.S.; Berglund, P. Biocatalytic promiscuity. Eur. J. Org. Chem. 2011, 3391–3401.
- Miao, Y.; Rahimi, M.; Geertsema, E.M.; Poelarends, G.J. Recent developments in enzyme promiscuity for carbon–carbon bond-forming reactions. Curr. Opin. Chem. Biol. 2015, 25, 115–123.
- Dwivedee, B.P.; Soni, S.; Sharma, M.; Bhaumik, J.; Laha, J.K.; Banerjee, U.C. Promiscuity of lipase-catalyzed reactions for organic synthesis: A recent update. ChemistrySelect 2018, 3, 2441–2466.
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505.
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 1985, 82, 3192–3196.
- Elgharbawya, A.A.; Riyadi, F.A.; Alama, M.d.Z.; Moniruzzaman, M. Ionic liquids as a potential solvent for lipase-catalysed reactions: A review. J. Mol. Liq. 2018, 251, 150–166.
- Tan, J.-N.; Dou, Y. Deep eutectic solvents for biocatalytic transformations: Focused lipase-catalyzed organic reactions. Appl. Microbiol. Biotechnol. 2020, 104, 1481–1496.
- Ghanem, A.; Aboul-Enein, H.Y. Application of lipases in kinetic resolution of racemates. Chirality 2005, 17, 1–15.
- Seddigi, Z.S.; Malik, M.S.; Ahmed, S.A.; Babalghith, A.O.; Kamal, A. Lipases in asymmetric transformations: Recent advances in classical kinetic resolution and lipase–metal combinations for dynamic processes. Coordin. Chem. Rev. 2017, 348, 54–70.
- Patti, A.; Sanfilippo, C. Breaking molecular symmetry through biocatalytic reactions to gain access to valuable chiral synthons. Symmetry 2020, 12, 1454.
- Thangaraj, B.; Solomon, P.R. Immobilization of lipases–A review Part II: Carrier Materials. ChemBioEng. Rev. 2019, 6, 67–194.
- Chandra, P.; Enespa, R.; Singh Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell. Fact. 2020, 19, 169.
- Kitazume, T.; Ikeya, T.; Murata, K. Synthesis of optically active trifluorinated compounds: Asymmetric Michael addition with hydrolytic enzymes. J. Chem. Soc. Chem. Commun. 1986, 1331–1333.
- Branneby, C.; Carlqvist, P.; Magnusson, A.; Hult, K.; Brinck, T.; Berglund, P. Carbon-carbon bonds by hydrolytic enzymes. J. Am. Chem. Soc. 2003, 125, 874–875.
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480.
- Li, C.; Feng, X.-W.; Wang, N.; Zhou, Y.-J.; Yu, X.-Q. Biocatalytic promiscuity: The first lipase-catalysed asymmetric aldol reaction. Green Chem. 2008, 10, 616–618.
- Xie, Z.-B.; Wang, N.; Zhou, L.-H.; Wan, F.; He, T.; Le, Z.-G.; Yu, X.-Q. Lipase-catalyzed stereoselective cross-aldol reaction promoted by water. ChemCatChem 2013, 5, 1935–1940.
- Xie, Z.-B.; Wang, N.; Jiang, G.-F.; Yu, X.-Q. Biocatalytic asymmetric aldol reaction in buffer solution. Tetrahedron Lett. 2013, 54, 945–948.
- Wang, Y.; Liang, X.-Y.; Chen, X.-Y.; Liang, Z.-H.; Cheng, H.; Li, X.; Li, L.-L. Promotion of water in BPL-catalyzed asymmetric cross aldol reaction. Biomass Convers. Biorefinery 2020.
- Wang, Y.; Cheng, H.; Li, X.; Li, L.-L.; Liang, Z.-H.; Liang, X.-Y.; Chen, X.-Y. MML-catalyzed direct aldol reaction in green solvents. Biomass Convers. Biorefinery 2020.
- Guan, Z.; Fu, J.-P.; He, Y.-H. Biocatalytic promiscuity: Lipase-catalyzed asymmetric aldol reaction of heterocyclic ketones with aldehydes. Tetrahedron Lett. 2012, 53, 4959–4961.
- Yıldız, T.; Yaşa, H.; Hasdemir, B.; Yusufoğlu, A.S. Different bio/Lewis acid-catalyzed stereoselective aldol reactions in various mediums. Monatsh. Chem. 2017, 148, 1445–1452.
- Zhang, Y.; Wang, N.; Xie, Z.-B.; Zhou, L.-H.; Yu, X.-Q. Ionic liquid as a recyclable and efficient medium for lipase-catalyzed asymmetric cross aldol reaction. J. Mol. Catal. B Enzym. 2014, 110, 100–110.
- González-Martínez, D.; Gotor, V.; Gotor-Fernández, V. Application of deep eutectic solvents in promiscuous lipase-catalysed aldol reactions. Eur. J. Org. Chem. 2016, 1513–1519.
- Branneby, C.; Carlqvist, P.; Hult, K.; Brinck, T.; Berglund, P. Aldol additions with mutant lipase: Analysis by experiments and theoretical calculations. J. Mol. Catal. B Enzym. 2004, 31, 123–128.
- Reznikov, A.N.; Klimochkin, Y.N. Recent developments in highly stereoselective Michael addition reactions catalyzed by metal complexes. Synthesis 2020, 52, 781–795.
- Malkar, R.S.; Jadhav, A.L.; Yadav, G.D. Innovative catalysis in Michael addition reactions for C-X bond formation. Mol. Catal. 2020, 485, 110814.
- Torre, O.; Alfonso, I.; Gotor, V. Lipase catalysed Michael addition of secondary amines to acrylonitrile. Chem. Commun. 2004, 1724–1725.
- Xu, J.-M.; Zhang, F.; Wu, Q.; Zhang, Q.-Y.; Lin, X.-F. Hydrolase-catalyzed Michael addition of 1,3-dicarbonyl compounds to α,β-unsaturated compounds in organic solvent. J. Mol. Catal. B Enzym. 2007, 49, 50–54.
- Steunenberg, P.; Sijm, M.; Zuilhof, H.; Sanders, J.P.M.; Scott, E.L.; Franssen, M.C.R. Lipase-catalyzed aza-Michael reaction on acrylate derivatives. J. Org. Chem. 2013, 78, 3802–3813.
- Rivera-Ramírez, J.D.; López-Munguía, J.E.A.; Castillo, A.M.E. Thermodynamically controlled chemoselectivity in lipase-catalyzed aza-Michael additions. J. Mol. Catal. B: Enzym. 2015, 112, 76–82.
- Strohmeier, G.A.; Sović, T.; Steinkellner, G.; Hartner, F.S.; Andryushkova, A.; Purkarthofer, T.; Glieder, A.; Gruber, K.; Griengl, H. Investigation of lipase-catalyzed Michael-type carbon–carbon bond formations. Tetrahedron 2009, 65, 5663–5668.
- Cai, J.-F.; Guan, Z.; He, Y.-H. The lipase-catalyzed asymmetric C–C Michael addition. J. Mol. Catal. B Enzym. 2011, 68, 240–244.
- Rizzo, P.V.S.; Boarin, L.A.; Freitas, I.O.M.; Gomes, R.S.; Beatriz, A.; Rinaldi, A.W.; Domingues, N.L.C. The study of biocatalyzed thio-Michael reaction: A greener and multi-gram protocol. Tetrahedron Lett. 2014, 55, 430–434.
- Zhou, L.-H.; Wang, N.; Chen, G.-N.; Yang, Q.; Yang, S.-Y.; Zhang, W.; Zhang, Y.; Yu, X.-Q. Lipase-catalyzed highly diastereoselective direct vinylogous Michael addition reaction of α,α-dicyanoolefins to nitroalkenes. J. Mol. Catal. B Enzym. 2014, 109, 170–177.
- Xie, B.-H.; Guan, Z.; He, Y.-H. Promiscuous enzyme-catalyzed Michael addition: Synthesis of warfarin and derivatives. J. Chem. Technol. Biotechnol. 2012, 87, 1709–1714.
- Sano, K.; Saito, S.; Hirose, Y.; Kohari, Y.; Nakano, H.; Seki, C.; Tokiwa, M.; Takeshita, M.; Uwai, K. Development of a novel method for warfarin synthesis via lipase-catalyzed steroselective Michael reaction. Heterocycles 2013, 87, 1269–1278.
- Fan, Y.; Cai, D.; Wang, X.; Yang, L. Ionic liquids: Efficient media for the lipase-catalyzed Michael addition. Molecules 2018, 23, 2154.
- Carlqvist, P.; Svedendahl, M.; Branneby, C.; Hult, K.; Brinck, T.; Berglund, P. Exploring the active-site of a rationally redesigned lipase for catalysis of Michael-type additions. ChemBioChem 2005, 6, 331–336.
- Gu, B.; Hu, Z.-E.; Yang, Z.-J.; Li, J.; Zhou, Z.-W.; Wang, N.; Yu, X.-Q. Probing the mechanism of CAL-B-catalyzed aza-Michael addition of aniline compounds with acrylates using mutation and molecular docking simulations. Chem. Sel. 2019, 4, 3848–3854.
- Li, K.; He, T.; Li, C.; Feng, X.-W.; Wang, N.; Yu, X.-Q. Lipase-catalysed direct Mannich reaction in water: Utilization of biocatalytic promiscuity for C–C bond formation in a “one-pot” synthesis. Green Chem. 2009, 11, 777–779.
- He, T.; Li, K.; Wu, M.-Y.; Feng, X.-W.; Wang, N.; Wang, H.-Y.; Li, C.; Yu, X.-Q. Utilization of biocatalytic promiscuity for direct Mannich reaction. J. Mol. Catal. B Enzym. 2010, 67, 189–194.
- Wu, L.-L.; Xiang, Y.; Yang, D.-C.; Guan, Z.; He, Y.-H. Biocatalytic asymmetric Mannich reaction of ketimines using wheat germ lipase. Catal. Sci. Technol. 2016, 6, 3963–3970.
- Ding, X.; Dong, C.-L.; Guan, Z.; He, Y.-H. Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: Direct enantioselective synthesis of 2,2-disubstituted indol-3-ones from 2-arylindoles. Angew. Chem. 2019, 131, 124–130.
- Xu, J.-C.; Li, W.-M.; Zheng, H.; Lai, Y.-F.; Zhang, P.-F. One-pot synthesis of tetrahydrochromene derivatives catalyzed by lipase. Tetrahedron 2011, 67, 9582–9587.
- Chen, X.; Zhang, W.; Yang, F.; Guo, C.; Zhao, Z.; Ji, D.; Zhou, F.; Wang, Z.; Zhao, R.; Wang, L. Synthesis of dihydropyranopyranes via a multi-component reaction catalyzed by lipase. Green Chem. Lett. Rev. 2017, 10, 54–58.
- Dalal, K.S.; Padvi, S.A.; Wagh, Y.B.; Dalal, D.S.; Chaudhari, B.L. Lipase from porcine pancreas: An efficient biocatalyst for the synthesis of ortho-aminocarbonitriles. Chem. Sel. 2018, 3, 10378–10382.
- Yang, Z.-J.; Gong, Q.-T.; Wang, Y.; Yu, Y.; Liu, Y.-H.; Wang, N.; Yu, X.-Q. Biocatalytic tandem multicomponent reactions for one-pot synthesis of 2-amino-4H-pyran library and in vitro biological evaluation. Mol. Catal. 2020, 491, 110983.
- Dalal, K.S.; Chaudhari, M.A.; Dalal, D.S.; Chaudhari, B.L. The first efficient biocatalytic route for the synthesis of Kojic acid derivatives in aqueous media. Catal. Comm. 2021, 152, 106289.
- Yin, D.-H.; Liu, W.; Wang, Z.-X.; Huang, X.; Zhang, J.; Huang, D.-C. Enzyme-catalyzed direct three-component aza-Diels–Alder reaction using lipase from Candida sp. 99–125. Chin. Chem. Lett. 2017, 28, 153–158.
- Wang, J.-L.; Chen, X.-Y.; Wu, Q.; Lin, X.-F. One-pot synthesis of spirooxazino derivatives via enzyme- initiated multicomponent reactions. Adv. Synth. Catal. 2014, 356, 999–1005.
- Chen, X.-Y.; Wang, J.-L.; Lin, X.-F.; Wu, Q. Lipase-initiated one-pot synthesis of spirooxazino derivatives: Redesign of multicomponent reactions to expand substrate scope and application potential. Tetrahedron 2016, 72, 3318–3323.
- Chen, X.-Y.; Hu, Y.-J.; Lin, X.-F.; Wu, Q. Asymmetric synthesis of strongly fluorescent spirooxazino derivatives via multi-enzymatic telescopic reactions. J. Mol. Catal. B Enzym. 2016, 133, S277–S280.

