 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chunsheng Kang | + 3335 word(s) | 3335 | 2020-12-29 03:09:56 | | | |
| 2 | Vivi Li | + 104 word(s) | 3439 | 2022-03-24 04:04:33 | | | | |
| 3 | Vivi Li | -2 word(s) | 3437 | 2022-03-25 07:47:39 | | |
Video Upload Options
The current understanding of miRNA biology is greatly derived from studies on the guide strands and the passenger strands, also called miRNAs*, which are considered as carriers with no sense for long periods. As such, various studies alter the expression of guide strands by manipulating the expression of their primary transcripts or precursors, both of which are premature miRNAs. In this situation, the regulatory miRNA* species may interfere with the phenotypic interpretation against the target miRNA. However, such methods could manipulate the expression of two functionally synergistic miRNAs of the same precursor, leading to therapeutic potential against various diseases, including cancers. Premature miRNAs represent an underappreciated target reservoir and provide molecular targets for “one-two punch” against cancers. Examples of targetable miRNA precursors and available targeting strategies are provided here.
1. Introduction
MicroRNA (miRNA) is a class of small nonprotein-coding RNAs (ncRNAs) that are 15–27-nucleotides (nt) in length, are ubiquitously expressed, and interact with most of the mammalian messenger RNAs (mRNAs) [1]. The interplay between miRNAs and mRNAs leads to decreased translational efficiency and/or mRNA levels, which is the predominant reason behind decreased protein production [2][3][4]. It is estimated that over 60% of mRNAs are conserved targets for endogenous miRNAs, and more mRNAs will be added to this conservative estimation when less conserved sites will be taken into account, indicating the existence of a previously unexpected post-transcriptional RNA signaling network [1]. Extensive screening for functional miRNAs has become easier due to the refinement of the principles that allow the manipulation of miRNA expression; functional miRNAs will gradually overturn the central dogma of molecular biology [5]—evidence regarding involvement of miRNAs in post-transcriptional regulation is mounting, and they are considered as the key components in the maintenance of intracellular homeostasis of many physiological processes [6][7]. Apart from their intracellular functions, miRNAs are also highlighted as unpredictable, important intercellular communicators, which help in the maintenance of a homeostatic extracellular microenvironment [8]. Dysregulated miRNAs in cell type-specific or cell-state-specific gene expression patterns cause pathophysiological transitions toward neurological [9], cardiovascular [10], metabolic [11], autoimmune [12], and carcinogenic disorders [13], and in turn, provide novel diagnostic, therapeutic, and prognostic opportunities in deciphering diseases [14].
2. Functional Annotation of Premature miRNAs in Cancers
The functional importance of miRNAs is being extensively studied. The functional role of miRNA* species in various diseases is highly valuable in annotating the functionality of miRNA precursors [15]. Almost all models of functional miRNAs, from diverse origins and biosynthesis pathways, converge at the step where a hairpin miRNA precursor is produced, and thus, functional annotation of premature miRNA is focused on the last premature form of miRNA, the pre-miRNA. Given the inability to establish direct contact with regulatory protein complexes, the functions of pre-miRNAs are determined by the joint activity of their encompassing regulatory miRNAs. In this entry, researchers have mainly focused on the pre-miRNAs that produce two mature strands and account for the majority of miRNA precursors. Mining functional information related to miRNA in combination with newly discovered biological implication of its passenger strand is essential for drafting the biological propensity of their parental precursor transcripts. In cancers, pre-miRNAs can be divided into three different subgroups, i.e., oncogenic, tumor suppressive, and ambivalent pre-miRNAs, based on the function of the dual sister strands (Figure 1a–c.).
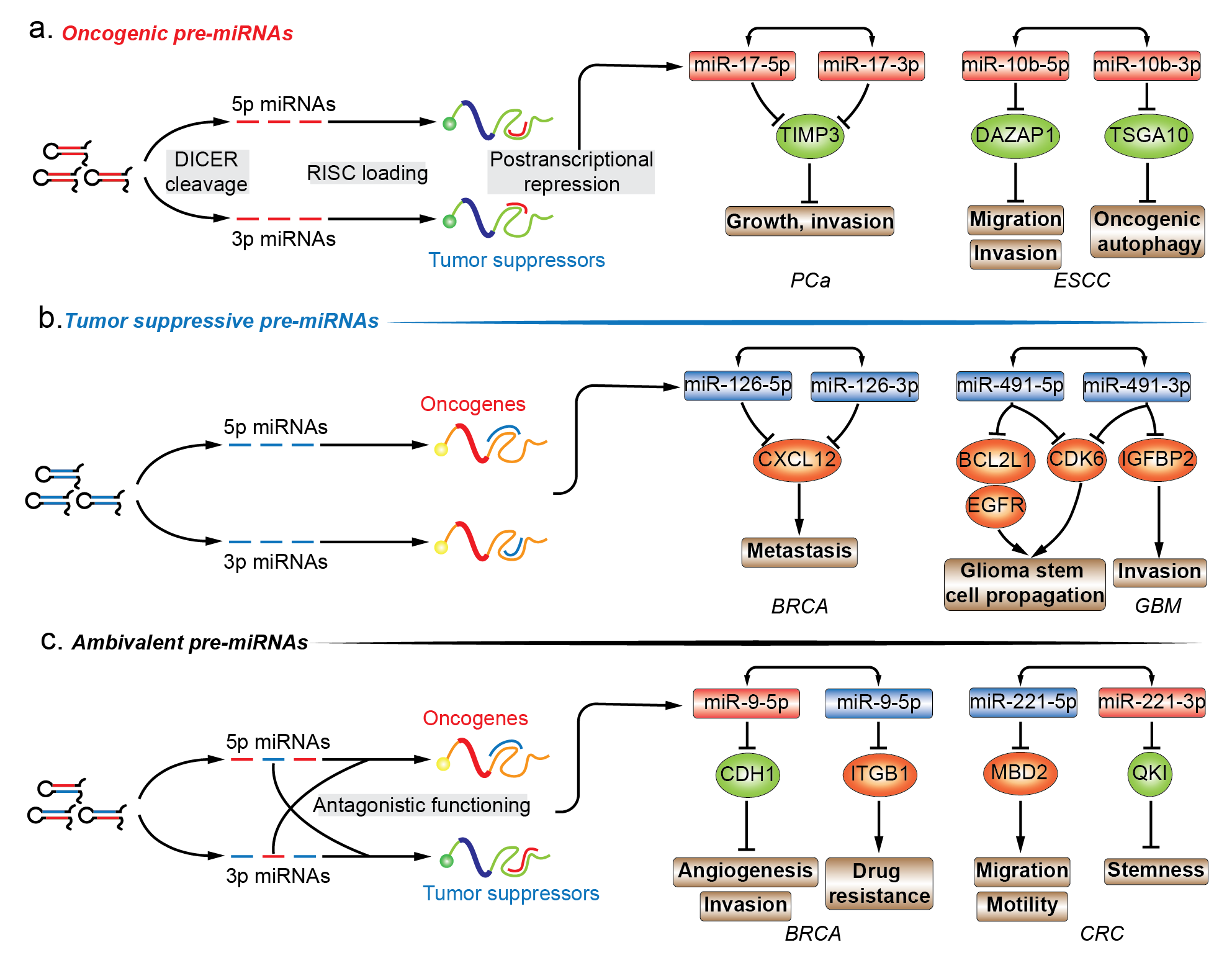
2.1. Oncogenic Pre-miRNAs
2.2. Tumor-Suppressive Pre-miRNAs
2.3. Ambivalent Pre-miRNAs
Unlike the aforementioned oncogenic or tumor suppressive pre-miRNA, ambivalent pre-miRNAs can be processed into two mature miRNAs which tend to function in two opposing directions (Figure 1c). For example, pre-miR-31 in oral squamous cell carcinoma (OSCC), pre-miR-9 and pre-miR-10b in BRCA consist of a single oncomiRs at the 5′ arm and tumor suppressive miRNA at the 3′ arm, while pre-miR-221 in colorectal cancer (CRC) are constituted in a reverse manner [35][36][37][38][39][40][41][42]. With respect to pre-miR-31, the oncogenic miR-31-5p in head and neck squamous cell carcinoma (HNSCC) were found to be one of the most highly expressed miRNAs when compared to normal tissues. The elevated miR-31-5p expression was linked to increased oncogenic potential of HNSCC cells by promoting cell growth and migration. The phenotypic effect of miR-31-5p was mediated by the suppression of factor-inhibiting hypoxia-inducible factor (FIH), which is the direct target of miR-31-5p. However, under normoxic conditions, FIH acts as a tumor suppressor by inhibiting oncogenicity [35]. The tumor suppressive complementary strand, miR-31-3p, was associated with decreased capability of growth and migration in SAS and Fadu OSCC cell lines. Ras homolog family member A (RHOA) was chosen as a candidate for observing the phenotypic changes in response to miR-31-3p manipulation and the miRNA-target relationship was verified by 3′-UTR luciferase reporter assays. Interestingly, the authors examined the phenotypic influence of forced pre-miR-31 expression, which supplemented two mature products in the form of oncogenic 5′ arm and tumor suppressive 3′ arm, and showed that the phenotypic profiling of ambivalent pre-miR-31 introduction was oncogenic [36]. Regarding pre-miR-9, oncogenic miR-9-5p was found to be significantly upregulated in human BRCA compared to normal breast tissues. Besides, c-myc induced an increase of more than 500-fold in the expression of miR-9-5p/3p in mice transgenic models [43][44]. In human cancer, miR-9-5p displayed a positive correlation with MYCN amplification and tumor grade neuroblastoma, and was elevated in patients with metastatic BRCA. In BRCA cells, 3′-UTR of cadherin 1 (CDH1) was directly targeted by miR-9-5p. CDH1 suppression derepressed β-catenin pathways and induced vascular endothelial growth factor A (VEGFA) expression levels, angiogenesis, mesenchymal transitions, and metastatic phenotypes in BRCA [37]. The pairing strand, viz., tumor suppressive miR-9-3p, was screened out from a miRNA mimics library for a possible synthetic sensitizer of MEK inhibitor, viz., AZD6244 in BRCA cell lines. In the presence of miR-9-3p, AZD6244-treated BRCA cells were subjected to vulnerable growth arrest, migration, and inhibition of invasion, which was mediated by miR-9-3p repression of integrin subunit beta 1 (ITGB1) [38]. With regards to pre-miR-10b, oncogenic miR-10b-5p was highly expressed in metastatic BRCA, and its upregulation in BRCA cell lines was competent enough to increase the cell motility and invasiveness in vitro and elicit tumor invasion and distant metastasis in vivo. The increase of miR-10b-5p was induced by TWIST1-binding to E-box located at the upstream of MIR10b stem-loop sequences, and the function was executed by direct targeting of 3′-UTR of homeoboxD10 (HOXD10). This is an established tumor suppressor system which impairs the migrating and invading abilities of BRCA cells in vivo [39][45]. The decreased miR-10b-3p expression was identified in BRCA tumor samples compared with matched peritumor tissue samples, indicating that the loss-of-function may contribute to tumorigenesis. In BRCA cell lines, ectopic introduction of miR-10b-3p inhibited cell viability in vitro and tumor growth in xenograft models. The possible molecular mechanism is the direct repression of the regulatory genes of cell cycle, viz., cyclin A2 (CCNA2), polo like kinase 1 (PLK1) and BUB1, whose expression levels were the most negatively correlated with miR-10b-3p in BRCA samples. The restoration of miR-10b-3p in BRCA cell lines increased subG1- and decreased S-phase cell populations by inhibiting dysregulated cell cycle-associated pathways, and all the three validated targets were associated with poor outcome in BRCA patients [40]. In contrast to the above-mentioned oncogenic 5′ arm-derived miRNA, tumor suppressive miR-221-5p were significantly downregulated in metastatic CRC cells, and its downregulation was associated with poor survival of CRC patients belonging to all the stages. Upregulation of miR-221-5p reduced the subcutaneous and orthotopic CRC tumor volumes in mice, and impaired migratory and invasive capability of CRC cells in vitro. Methyl-CpG binding domain protein 2 (MBD2) contains a validated binding site for miR-221-5p, and its knockdown recaptured the phenotypic change of miR-221-5p introduction in CRC cells [41]. The oncogenic strand of pre-miR-221 in human CRC, miR-221-3p, was preferentially overexpressed in CRC stem cell-like cells, and its overexpression correlated with the reduced survivability in CRC patients. Overexpressed miR-221-3p promoted the three-dimensional (3D) organoid-forming capacity of human CRC cells in vitro, and miR-221-3p antagonization repressed in vivo engraftment and growth of human CRC patient-derived xenografts (PDX) in mice models. QKI-5 was experimentally validated to be the functional target of miR-221-3p, which impeded the 3D-organoid formation and tumorigenic capability of human CRC PDX cells [42]. In certain cellular backgrounds, therapeutic manipulation of ambivalent pre-miRNAs may be infeasible as the expression of the dual sister strands are inevitably interfered simultaneously and unidirectionally, which leads to transcriptional repression of oncogenes as well as tumor suppressors.
3. Conclusion and Future Perspective
Without any doubt, miRNA species have grown to be one of the most important regulatory molecules in maintaining intracellular hemostasis. The evolving knowledge about the biogenesis and function of miRNAs keeps illuminating miRNA biology through unprecedented depth and breadth. The early stage of functional evaluation of miRNA species is focused mostly on the guide strands of miRNA duplex, and the passenger strand is considered to be nonsense and degraded after the guide strand has been loaded into the RISC. With the increase in evidence concerning substantial regulatory activities of miRNA*, the nonsense tag of passenger strand has been gradually torn off. As a consequence, some of the currently available mature miRNA-targeting strategies have been deemed less rigorous in cancer research due to the influence of nonvoluntary negligence in miRNA* expressions. Reviewing the various biogenesis pathways leading to functional miRNAs, impertinence with respect to these inappropriately applied strategies converge on the attempt to manipulate a single miRNA at premature stages. This should be avoided such that simultaneously altered dual strands could either mitigate or exaggerate the performance of the target miRNA-mediated phenotype. One of the major advantages of premature miRNA manipulation rises from the lesson of utilizing such methodology in single miRNA manipulation. If the dual sister strands function synergistically, it would not be more appropriate to use the same strategies for exaggerating the therapeutic performance. The theory of “target-two-sets-of-genes-with-one-pre-miRNA” has been proposed. Currently, only a very small portion of studies are dedicated to establishing the functional relevance of dual sister strands in one study. Functional annotation of a pre-miRNA based on multiple separated studies might face a conflicted situation that ambivalent functions of one of the mature strands are reported. A problem is that the investigations of dual sister strands of a miRNA precursor are mostly not launched in the same lab at the same period of time. It is inevitable that a contradicted phenotype occurs due to the different experimental conditions. However, it can be anticipated that in company with the developing understanding of miRNA* species, functional annotation of pre-miRNA will be taken into account, and annotated pre-miRNA-based therapeutic will sprout with less controversy. Theoretically, a pre-miRNA should be functionally annotated in a single study to rule out the experimental bias, and ideally, phenotypical profiling should be performed before therapeutic interventions. The phenotypic profiling of dual regulatory strands may grant superior performance to the therapeutic interventions on certain premature miRNA, which acts as a “one-two punch” strategy against malignancies. The functional annotation of a miRNA precursor will be generously aided by the availability of annotated miRNA species. Researchers envision that in the upcoming future, there will be a significant decrease in the inappropriate utilization of premature miRNA inventions on single miRNA manipulations and generous increase in the number of functional miRNA precursor annotations. Being an almost indispensable step of miRNA maturation, the miRNA precursor may deserve a functional tag in the future, acting as a molecular target instead of a mere carrier of regulatory miRNA/miRNA*.
References
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105.
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110.
- Iwakawa, H.; Tomari, Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015, 25, 651–665.
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840.
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433.
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531.
- Ebert, M.S.; Sharp, P.A. Roles for MicroRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524.
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. Secreted microRNAs: A new form of intercellular communication. Trends Cell Biol. 2012, 22, 125–132.
- Kosik, K.S. The neuronal microRNA system. Nat. Rev. Neurosci. 2006, 7, 911–920.
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342.
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–251.
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122.
- Berindan-Neagoe, I.; Monroig, P.D.C.; Pasculli, B.; Calin, G.A. MicroRNAome genome: A treasure for cancer diagnosis and therapy. CA Cancer J. Clin. 2014, 64, 311–336.
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221.
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162.
- Yang, X.; Du, W.W.; Li, H.; Liu, F.; Khorshidi, A.; Rutnam, Z.J.; Yang, B.B. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 2013, 41, 9688–9704.
- Shan, S.W.; Fang, L.; Shatseva, T.; Rutnam, Z.J.; Yang, X.; Du, W.; Lu, W.-Y.; Xuan, J.W.; Deng, Z.; Yang, B.B. Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. J. Cell Sci. 2013, 126, 1517–1530.
- Chen, Y.; Lu, Y.; Ren, Y.; Yuan, J.; Zhang, N.; Kimball, H.; Zhou, L.; Yang, M. Starvation-induced suppression of DAZAP1 by miR-10b integrates splicing control into TSC2-regulated oncogenic autophagy in esophageal squamous cell carcinoma. Theranostics 2020, 10, 4983–4996.
- Zhang, Q.; Zhang, J.; Fu, Z.; Dong, L.; Tang, Y.; Xu, C.; Wang, H.; Zhang, T.; Wu, Y.; Dong, C.; et al. Hypoxia-induced microRNA-10b-3p promotes esophageal squamous cell carcinoma growth and metastasis by targeting TSGA10. Aging 2019, 11, 10374–10384.
- Hirata, Y.; Murai, N.; Yanaihara, N.; Saito, M.; Saito, M.; Urashima, M.; Murakami, Y.; Matsufuji, S.; Okamoto, A. MicroRNA-21 is a candidate driver gene for 17q23-25 amplification in ovarian clear cell carcinoma. BMC Cancer 2014, 14, 799.
- Lou, Y.; Yang, X.; Wang, F.; Cui, Z.; Huang, Y. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int. J. Mol. Med. 2010, 26, 521–527.
- Pink, R.C.; Samuel, P.; Massa, D.; Caley, D.P.; Brooks, S.A.; Carter, D.R.F. The passenger strand, miR-21-3p, plays a role in mediating cisplatin resistance in ovarian cancer cells. Gynecol. Oncol. 2015, 137, 143–151.
- Shao, N.; Ma, G.; Zhang, J.; Zhu, W. MIR-221-5p enhances cell proliferation and metastasis through post-transcriptional regulation of SOCS1 in human prostate cancer. BMC Urol. 2018, 18, 1–9.
- Sun, T.; Wang, X.; He, H.H.; Sweeney, C.J.; Liu, S.X.; Brown, M.; Balk, S.; Lee, G.S.; Kantoff, P.W. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene 2014, 33, 2790–2800.
- Chan, J.K.; Blansit, K.; Kiet, T.; Sherman, A.; Wong, G.; Earle, C.; Bourguignon, L.Y.W. The inhibition of miR-21 promotes apoptosis and chemosensitivity in ovarian cancer. Gynecol. Oncol. 2014, 132, 739–744.
- Javanmardi, S.; Tamaddon, A.M.; Aghamaali, M.R.; Ghahramani, L.; Abolmaali, S.S. Redox-sensitive, PEG-shielded carboxymethyl PEI nanogels silencing MicroRNA-21, sensitizes resistant ovarian cancer cells to cisplatin. Asian J. Pharm. Sci. 2020, 15, 69–82.
- Echevarría-Vargas, I.M.; Valiyeva, F.; Vivas-Mejía, P.E. Upregulation of miR-21 in cisplatin resistant ovarian cancer via JNK-1/c-Jun pathway. PLoS ONE 2014, 9.
- Zhang, X.Y.; Li, Y.F.; Ma, H.; Gao, Y.H. Regulation of MYB mediated cisplatin resistance of ovarian cancer cells involves miR-21-wnt signaling axis. Sci. Rep. 2020, 10, 1–8.
- Sun, T.; Wang, Q.; Balk, S.; Brown, M.; Lee, G.S.M.; Kantoff, P. The role of microrna-221 and microrna-222 in Androgen- independent prostate cancer cell lines. Cancer Res. 2009, 69, 3356–3363.
- Zhang, Y.; Yang, P.; Sun, T.; Li, D.; Xu, X.; Rui, Y.; Li, C.; Chong, M.; Ibrahim, T.; Mercatali, L.; et al. MiR-126 and miR-126 * repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat. Cell Biol. 2013, 15, 284–294.
- Uchida, A.; Seki, N.; Mizuno, K.; Misono, S.; Yamada, Y.; Kikkawa, N.; Sanada, H.; Kumamoto, T.; Suetsugu, T.; Inoue, H. Involvement of dual-strand of the miR-144 duplex and their targets in the pathogenesis of lung squamous cell carcinoma. Cancer Sci. 2019, 110, 420–432.
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017, 406, 12–21.
- Li, X.; Liu, Y.; Granberg, K.J.; Wang, Q.; Moore, L.M.; Ji, P.; Gumin, J.; Sulman, E.P.; Calin, G.A.; Haapasalo, H.; et al. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene 2015, 34, 1619–1628.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Liu, C.J.; Tsai, M.M.; Hung, P.S.; Kao, S.Y.; Liu, T.Y.; Wu, K.J.; Chiou, S.H.; Lin, S.C.; Chang, K.W. miR-31 ablates expression of the HIF regulatory factor FIH to activate the HIF pathway in head and neck carcinoma. Cancer Res. 2010, 70, 1635–1644.
- Chang, K.W.; Kao, S.Y.; Wu, Y.H.; Tsai, M.M.; Tu, H.F.; Liu, C.J.; Lui, M.T.; Lin, S.C. Passenger strand miRNA miR-31 regulates the phenotypes of oral cancer cells by targeting RhoA. Oral Oncol. 2013, 49, 27–33.
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. MiR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256.
- Zawistowski, J.S.; Nakamura, K.; Parker, J.S.; Granger, D.A.; Golitz, B.T.; Johnson, G.L. MicroRNA 9-3p targets 1 integrin to sensitize claudin-low breast cancer cells to MEK inhibition. Mol. Cell. Biol. 2013, 33, 2260–2274.
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688.
- Biagioni, F.; Bossel Ben-Moshe, N.; Fontemaggi, G.; Canu, V.; Mori, F.; Antoniani, B.; Di Benedetto, A.; Santoro, R.; Germoni, S.; De Angelis, F.; et al. miR-10b *, a master inhibitor of the cell cycle, is down-regulated in human breast tumours. EMBO Mol. Med. 2012, 4, 1214–1229.
- Yuan, K.; Xie, K.; Fox, J.; Zeng, H.; Gao, H.; Huang, C.; Wu, M. Decreased levels of mir-224 and the passenger strand of miR-221 increase MBD2, suppressing maspin and promoting colorectal tumor growth and metastasis in mice. Gastroenterology 2013, 145, 853–864.e9.
- Mukohyama, J.; Isobe, T.; Hu, Q.; Hayashi, T.; Watanabe, T.; Maeda, M.; Yanagi, H.; Qian, X.; Yamashita, K.; Minami, H.; et al. MiR-221 targets QKI to enhance the tumorigenic capacity of human colorectal cancer stem cells. Cancer Res. 2019, 79, 5151–5158.
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070.
- Sun, Y.; Wu, J.; Wu, S.H.; Thakur, A.; Bollig, A.; Huang, Y.; Joshua Liao, D. Expression profile of microRNAs in c-Myc induced mouse mammary tumors. Breast Cancer Res. Treat. 2009, 118, 185–196.
- Carrio, M.; Arderiu, G.; Myers, C.; Boudreau, N.J. Homeobox D10 induces phenotypic reversion of breast tumor cells in a three-dimensional culture model. Cancer Res. 2005, 65, 7177–7185.

