 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Volatier | + 2795 word(s) | 2795 | 2022-02-18 05:01:48 | | | |
| 2 | Lindsay Dong | Meta information modification | 2795 | 2022-03-23 02:06:40 | | |
Video Upload Options
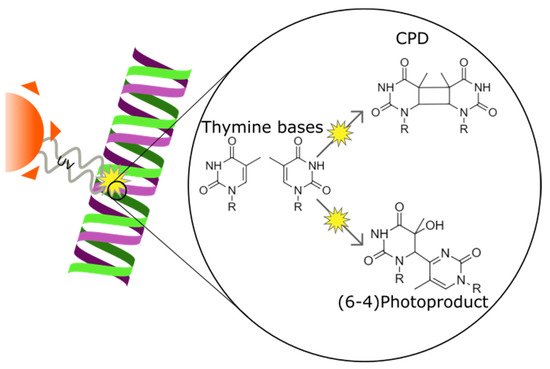
Ultraviolet (UV) irradiation induces DNA lesions in all directly exposed tissues. In the human body, two tissues are chronically exposed to UV: the skin and the cornea. The most frequent UV-induced DNA lesions are cyclobutane pyrimidine dimers (CPDs) that can lead to apoptosis or induce tumorigenesis. Lacking the protective pigmentation of the skin, the transparent cornea is particularly dependent on nucleotide excision repair (NER) to remove UV-induced DNA lesions. The DNA damage response also triggers intracellular autophagy mechanisms to remove damaged material in the cornea. Therapeutic solutions involving xenogenic DNA-repair enzymes such as T4 endonuclease V or photolyases exist and are widely distributed for dermatological use.
1. Cornea
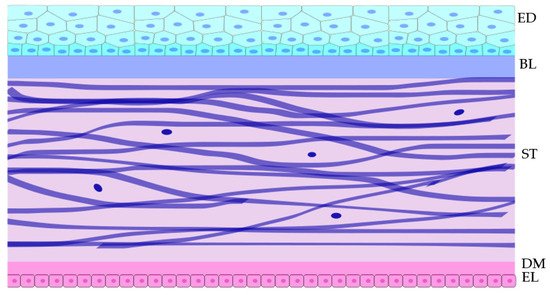
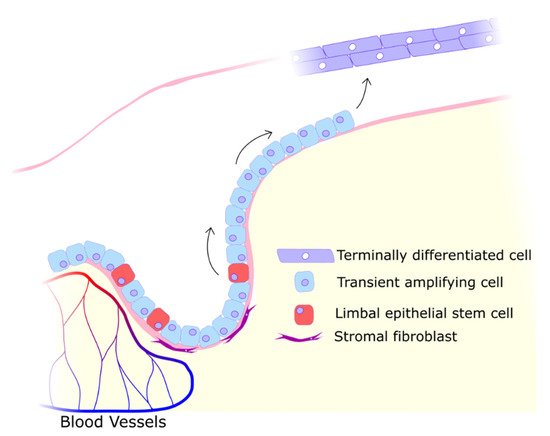
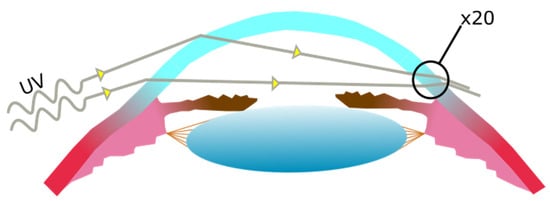
2. UV Effects on the Cornea
2.1. UV Damage and Repair Mechanisms
In the case of the cornea, acute UV exposure is known to cause photokeratitis, a painful inflammation where the epithelium, stroma, and endothelium may be affected and which leads to clouding [15][16][17][18]. Chronic UV exposure usually leads to long term conditions such as tumours (squamous cell carcinomas, malignant melanomas, lymphoma of the conjunctiva [19][20][21]) or keratopathy [22][23][24].
2.2. Pterygium Aetiology and Pathogenesis
2.3. UV-Induced DNA Lesion Formation
2.4. The Role of Genotoxic Stress
2.5. Reactive Oxygen Species
2.6. Disruption of Autophagy Mechanisms
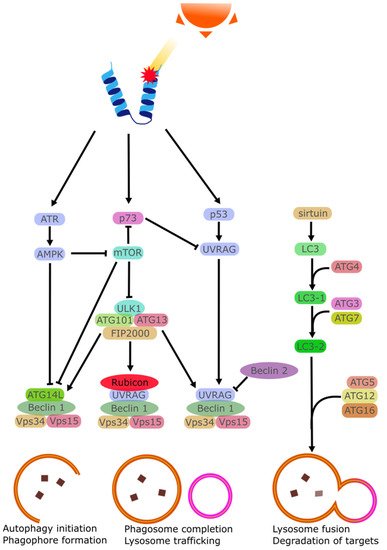
2.7. Apoptosis
Apoptosis can be triggered by several factors such as cytokines, hormones, virus, or drugs. At the right concentrations, these stimuli lead to the activation of effector caspases that trigger the apoptotic program [76]. UV irradiation has been shown to cause the activation of ATP-sensitive potassium (K(ATP)) channels in affected cells causing a loss of K+ ions and a depolarization of the mitochondrial membrane [77][78]. Intracellular K+ is importantly linked with the inhibition of caspase, and K+ loss is linked with greater rates of apoptosis [79]. In the cornea specifically, it has been observed that while in vivo cells did not perish following exposure to background doses of UVB, cultured cells did perish when exposed to similar UVB doses [80]. It was found that tear film provided a high amount of K+ ions to the corneal epithelia, ensuring a degree of apoptosis-resistance to the chronically UV-irradiated epithelial cells [81]. This is in contrast with the skin which lacks this excess K+ and instead regularly sees UV-triggered apoptosis in the form of sunburn cells [82][83].
3. UV Pathogenesis and Rescue
References
- DelMonte, D.W.; Kim, T. Anatomy and physiology of the cornea. J. Cataract Refract. Surg. 2011, 37, 588–598.
- Maurice, D.M. The structure and transparency of the cornea. J. Physiol. 1957, 136, 263–286.
- Schwarz, W.; Keyserlingk, D.G. On the fine structure of the human cornea with special reference to the problem of transparency. Z. Zellforsch Mikrosk. Anat. 1966, 73, 540–548.
- Kruse, F.E. Stem cells and corneal epithelial regeneration. Eye 1994, 8, 170–183.
- Zieske, J.D. Perpetuation of stem cells in the eye. Eye 1994, 8, 163–169.
- Yoon, J.J.; Ismail, S.; Sherwin, T. Limbal stem cells: Central concepts of corneal epithelial homeostasis. World J. Stem Cells 2014, 6, 391–403.
- Notara, M.; Behoudifard, S.; S. Kluth, M.A.; Masslo, C.; Ganss, C.; Frank, M.H.; Cursiefen, C. UV light-blocking contact lenses prevent UVB-induced DNA and oxidative damage of the limbal stem cell niche, protect against inflammation and maintain putative stem cell phenotype. Investig. Ophthalmol. Vis. Sci. 2019, 60, 920.
- Notara, M.; Refaian, N.; Brown, G.; Steven, P.; Bock, F.; Cursiefen, C. Effects of UVB irradiation on limbal stem cell niche and its role in cornea lymphangiogenesis. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5622.
- Notara, M.; Refaian, N.; Braun, G.; Steven, P.; Bock, F.; Cursiefen, C. Short-Term Ultraviolet A Irradiation Leads to Dysfunction of the Limbal Niche Cells and an Antilymphangiogenic and Anti-inflammatory Micromilieu. Investig. Ophthalmol. Vis. Sci. 2016, 57, 928–939.
- Notara, M.; Braun, G.; Dreisow, M.L.; Bock, F.; Cursiefen, C. Avastin effects on human limbal epithelial cell function and phenotype in vitro. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4343.
- Gao, X.; Guo, K.; Santosa, S.M.; Montana, M.; Yamakawa, M.; Hallak, J.A.; Han, K.Y.; Doh, S.J.; Rosenblatt, M.I.; Chang, J.H.; et al. Application of corneal injury models in dual fluorescent reporter transgenic mice to understand the roles of the cornea and limbus in angiogenic and lymphangiogenic privilege. Sci. Rep. 2019, 9, 12331.
- Azar, D.T. Corneal angiogenic privilege: Angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2006, 104, 264–302.
- Klyce, S.D. 12 Endothelial pump and barrier function. Exp. Eye Res. 2020, 198, 108068.
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45.
- Kronschlager, M.; Talebizadeh, N.; Yu, Z.; Meyer, L.M.; Lofgren, S. Apoptosis in Rat Cornea After In Vivo Exposure to Ultraviolet Radiation at 300 nm. Cornea 2015, 34, 945–949.
- Miller, S. A focus on ultraviolet keratitis. Nursing 2009, 4, 12–16.
- Najjar, D.M.; Awwad, S.T.; Zein, W.M.; Haddad, W.F. Assessment of the corneal endothelium in acute ultraviolet keratitis. Med. Sci. Monit. 2006, 12, MT23–MT25.
- Willmann, G. Ultraviolet Keratitis: From the Pathophysiological Basis to Prevention and Clinical Management. High Alt. Med. Biol. 2015, 16, 277–282.
- Coroi, M.C.; Rosca, E.; Mutiu, G.; Coroi, T. Squamous carcinoma of the conjunctiva. Rom. J. Morphol. Embryol. 2011, 52 (Suppl. 1), 513–515.
- Olah, Z. Malignant tumor of the cornea and exroderma pigmentosum. Ceskoslovenska Oftalmol. 1968, 24, 119–122.
- Toshida, H.; Nakayasu, K.; Okisaka, S.; Kanai, A. Incidence of tumors and tumor-like lesions in the conjunctiva and the cornea. Nippon Ganka Gakkai Zasshi 1995, 99, 186–189.
- Newton, R.; Ferlay, J.; Reeves, G.; Beral, V.; Parkin, D.M. Effect of ambient solar ultraviolet radiation on incidence of squamous-cell carcinoma of the eye. Lancet 1996, 347, 1450–1451.
- Hatsusaka, N.; Yamamoto, N.; Miyashita, H.; Shibuya, E.; Mita, N.; Yamazaki, M.; Shibata, T.; Ishida, H.; Ukai, Y.; Kubo, E.; et al. Association among pterygium, cataracts, and cumulative ocular ultraviolet exposure: A cross-sectional study in Han people in China and Taiwan. PLoS ONE 2021, 16, e0253093.
- Sekelj, S.; Dekaris, I.; Kondza-Krstonijevic, E.; Gabric, N.; Predovic, J.; Mitrovic, S. Ultraviolet light and pterygium. Coll. Antropol. 2007, 31 (Suppl. 1), 45–47.
- Coroneo, M.T.; Muller-Stolzenburg, N.W.; Ho, A. Peripheral light focusing by the anterior eye and the ophthalmohelioses. Ophthalmic. Surg. 1991, 22, 705–711.
- Dushku, N.; Reid, T.W. Immunohistochemical evidence that human pterygia originate from an invasion of vimentin-expressing altered limbal epithelial basal cells. Curr. Eye Res. 1994, 13, 473–481.
- Hill, J.C.; Maske, R. Pathogenesis of pterygium. Eye 1989, 3, 218–226.
- Fonseca, E.C.; Rocha, E.M.; Arruda, G.V. Comparison among adjuvant treatments for primary pterygium: A network meta-analysis. Br. J. Ophthalmol. 2018, 102, 748–756.
- Lu, P.; Chen, X.; Kang, Y.; Ke, L.; Wei, X.; Zhang, W. Pterygium in Tibetans: A population-based study in China. Clin. Exp. Ophthalmol. 2007, 35, 828–833.
- Stevenson, L.J.; Mackey, D.A.; Lingham, G.; Burton, A.; Brown, H.; Huynh, E.; Tan, I.J.; Franchina, M.; Sanfilippo, P.G.; Yazar, S. Has the Sun Protection Campaign in Australia Reduced the Need for Pterygium Surgery Nationally? Ophthalmic. Epidemiol. 2021, 28, 105–113.
- Hirst, L.W.; Smith, J. Accuracy of diagnosis of pterygium by optometrists and general practitioners in Australia. Clin. Exp. Optom. 2020, 103, 197–200.
- Khanna, R.C.; Marmamula, S.; Cicinelli, M.V.; Mettla, A.L.; Giridhar, P.; Banerjee, S.; Shekhar, K.; Chakrabarti, S.; Murthy, G.V.S.; Gilbert, C.E.; et al. Fifteen-year incidence rate and risk factors of pterygium in the Southern Indian state of Andhra Pradesh. Br. J. Ophthalmol. 2021, 105, 619–624.
- Fang, X.L.; Chong, C.C.Y.; Thakur, S.; Da Soh, Z.; Teo, Z.L.; Majithia, S.; Lim, Z.W.; Rim, T.H.; Sabanayagam, C.; Wong, T.Y.; et al. Ethnic differences in the incidence of pterygium in a multi-ethnic Asian population: The Singapore Epidemiology of Eye Diseases Study. Sci. Rep. 2021, 11, 501.
- Rim, T.H.; Kang, M.J.; Choi, M.; Seo, K.Y.; Kim, S.S. The incidence and prevalence of pterygium in South Korea: A 10-year population-based Korean cohort study. PLoS ONE 2017, 12, e0171954.
- Kwok, L.S.; Kuznetsov, V.A.; Ho, A.; Coroneo, M.T. Prevention of the adverse photic effects of peripheral light-focusing using UV-blocking contact lenses. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1501–1507.
- Sage, E. Distribution and repair of photolesions in DNA: Genetic consequences and the role of sequence context. Photochem. Photobiol. 1993, 57, 163–174.
- Rochette, P.J.; Bastien, N.; Todo, T.; Drouin, R. Pyrimidine (6-4) pyrimidone photoproduct mapping after sublethal UVC doses: Nucleotide resolution using terminal transferase-dependent PCR. Photochem. Photobiol. 2006, 82, 1370–1376.
- Liu, Z.; Tan, C.; Guo, X.; Kao, Y.T.; Li, J.; Wang, L.; Sancar, A.; Zhong, D. Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc. Natl. Acad. Sci. USA 2011, 108, 14831–14836.
- Torizawa, T.; Ueda, T.; Kuramitsu, S.; Hitomi, K.; Todo, T.; Iwai, S.; Morikawa, K.; Shimada, I. Investigation of the cyclobutane pyrimidine dimer (CPD) photolyase DNA recognition mechanism by NMR analyses. J. Biol. Chem. 2004, 279, 32950–32956.
- Hutchinson, F. Induction of tandem-base change mutations. Mutat. Res. 1994, 309, 11–15.
- Yamada, D.; Dokainish, H.M.; Iwata, T.; Yamamoto, J.; Ishikawa, T.; Todo, T.; Iwai, S.; Getzoff, E.D.; Kitao, A.; Kandori, H. Functional Conversion of CPD and (6-4) Photolyases by Mutation. Biochemistry 2016, 55, 4173–4183.
- Mees, A.; Klar, T.; Gnau, P.; Hennecke, U.; Eker, A.P.; Carell, T.; Essen, L.O. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 2004, 306, 1789–1793.
- McGregor, W.G.; Chen, R.H.; Lukash, L.; Maher, V.M.; McCormick, J.J. Cell cycle-dependent strand bias for UV-induced mutations in the transcribed strand of excision repair-proficient human fibroblasts but not in repair-deficient cells. Mol. Cell Biol. 1991, 11, 1927–1934.
- Steurer, B.; Turkyilmaz, Y.; van Toorn, M.; van Leeuwen, W.; Escudero-Ferruz, P.; Marteijn, J.A. Fluorescently-labelled CPD and 6-4PP photolyases: New tools for live-cell DNA damage quantification and laser-assisted repair. Nucleic Acids Res. 2019, 47, 3536–3549.
- Li, Y.F.; Kim, S.T.; Sancar, A. Evidence for lack of DNA photoreactivating enzyme in humans. Proc. Natl. Acad. Sci. USA 1993, 90, 4389–4393.
- van der Spek, P.J.; Kobayashi, K.; Bootsma, D.; Takao, M.; Eker, A.P.; Yasui, A. Cloning, tissue expression, and mapping of a human photolyase homolog with similarity to plant blue-light receptors. Genomics 1996, 37, 177–182.
- Munoz, M.J.; Nieto Moreno, N.; Giono, L.E.; Cambindo Botto, A.E.; Dujardin, G.; Bastianello, G.; Lavore, S.; Torres-Mendez, A.; Menck, C.F.M.; Blencowe, B.J.; et al. Major Roles for Pyrimidine Dimers, Nucleotide Excision Repair, and ATR in the Alternative Splicing Response to UV Irradiation. Cell Rep. 2017, 18, 2868–2879.
- Hsu, P.H.; Hanawalt, P.C.; Nouspikel, T. Nucleotide excision repair phenotype of human acute myeloid leukemia cell lines at various stages of differentiation. Mutat. Res.-Fund. Mol. Mech. Mutagenesis 2007, 614, 3–15.
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481.
- Langie, S.A.; Wilms, L.C.; Hamalainen, S.; Kleinjans, J.C.; Godschalk, R.W.; van Schooten, F.J. Modulation of nucleotide excision repair in human lymphocytes by genetic and dietary factors. Br. J. Nutr. 2010, 103, 490–501.
- de Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460.
- Vermeulen, W.; de Boer, J.; Citterio, E.; van Gool, A.J.; van der Horst, G.T.; Jaspers, N.G.; de Laat, W.L.; Sijbers, A.M.; van der Spek, P.J.; Sugasawa, K.; et al. Mammalian nucleotide excision repair and syndromes. Biochem. Soc. Trans. 1997, 25, 309–315.
- Rapin, I. Disorders of nucleotide excision repair. Handb. Clin. Neurol. 2013, 113, 1637–1650.
- Mallet, J.D.; Dorr, M.M.; Drigeard Desgarnier, M.C.; Bastien, N.; Gendron, S.P.; Rochette, P.J. Faster DNA Repair of Ultraviolet-Induced Cyclobutane Pyrimidine Dimers and Lower Sensitivity to Apoptosis in Human Corneal Epithelial Cells than in Epidermal Keratinocytes. PLoS ONE 2016, 11, e0162212.
- Mann, A.; Tighe, B. Contact lens interactions with the tear film. Exp. Eye Res. 2013, 117, 88–98.
- Wang, S.; Jiang, B.; Gu, Y. Changes of tear film function after pterygium operation. Ophthalmic. Res. 2011, 45, 210–215.
- Poljsak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012, 2012, 135206.
- de Jager, T.L.; Cockrell, A.E.; Du Plessis, S.S. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Adv. Exp. Med. Biol. 2017, 996, 15–23.
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424.
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158.
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209.
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627.
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112.
- Liu, M.; Zeng, T.; Zhang, X.; Liu, C.; Wu, Z.; Yao, L.; Xie, C.; Xia, H.; Lin, Q.; Xie, L.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139.
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell Biol. 1984, 4, 1689–1694.
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814.
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100.
- Zeng, R.; Chen, Y.; Zhao, S.; Cui, G.H. Autophagy counteracts apoptosis in human multiple myeloma cells exposed to oridonin in vitro via regulating intracellular ROS and SIRT1. Acta Pharmacol. Sin. 2012, 33, 91–100.
- Garva, R.; Thepmalee, C.; Yasamut, U.; Sudsaward, S.; Guazzelli, A.; Rajendran, R.; Tongmuang, N.; Khunchai, S.; Meysami, P.; Limjindaporn, T.; et al. Sirtuin Family Members Selectively Regulate Autophagy in Osteosarcoma and Mesothelioma Cells in Response to Cellular Stress. Front. Oncol. 2019, 9, 949.
- Shamsher, E.; Guo, L.; Davis, B.M.; Luong, V.; Ravindran, N.; Somavarapu, S.; Cordeiro, M.F. Resveratrol nanoparticles are neuroprotective in a rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2423.
- Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Resveratrol and Ophthalmic Diseases. Nutrients 2016, 8, 200.
- Tsai, T.Y.; Chen, T.C.; Wang, I.J.; Yeh, C.Y.; Su, M.J.; Chen, R.H.; Tsai, T.H.; Hu, F.R. The Effect of Resveratrol on Protecting Corneal Epithelial Cells from Cytotoxicity Caused by Moxifloxacin and Benzalkonium Chloride. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1575–1584.
- Ma, S.S.; Yu, Z.; Feng, S.F.; Chen, H.J.; Chen, H.Y.; Lu, X.H. Corneal autophagy and ocular surface inflammation: A new perspective in dry eye. Exp. Eye Res. 2019, 184, 126–134.
- Van Acker, S.I.; van den Bogerd, B.; Haagdorens, M.; Siozopoulou, V.; Dhubhghaill, S.N.; Pintelon, I.; Koppen, C. Pterygium-The Good, the Bad, and the Ugly. Cells 2021, 10, 1567.
- Martin, L.M.; Jeyabalan, N.; Tripathi, R.; Panigrahi, T.; Johnson, P.J.; Ghosh, A.; Mohan, R.R. Autophagy in corneal health and disease: A concise review. Ocul. Surf. 2019, 17, 186–197.
- Cao, C.; Healey, S.; Amaral, A.; Lee-Couture, A.; Wan, S.; Kouttab, N.; Chu, W.; Wan, Y. ATP-sensitive potassium channel: A novel target for protection against UV-induced human skin cell damage. J. Cell Physiol. 2007, 212, 252–263.
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228.
- Tang, J.Y.; Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell 2000, 5, 737–744.
- Hughes, F.M., Jr.; Bortner, C.D.; Purdy, G.D.; Cidlowski, J.A. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J. Biol. Chem. 1997, 272, 30567–30576.
- Singleton, K.R.; Will, D.S.; Schotanus, M.P.; Haarsma, L.D.; Koetje, L.R.; Bardolph, S.L.; Ubels, J.L. Elevated extracellular K+ inhibits apoptosis of corneal epithelial cells exposed to UV-B radiation. Exp. Eye Res. 2009, 89, 140–151.
- Leerar, J.R.; Glupker, C.D.; Schotanus, M.P.; Ubels, J.L. The effect of K(+) on caspase activity of corneal epithelial cells exposed to UVB. Exp. Eye Res. 2016, 151, 23–25.
- Teraki, Y.; Shiohara, T. Apoptosis and the skin. Eur. J. Dermatol. 1999, 9, 413–425, quiz 426.
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768.
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704.
- Vechtomova, Y.L.; Telegina, T.A.; Kritsky, M.S. Evolution of Proteins of the DNA Photolyase/Cryptochrome Family. Biochemistry 2020, 85, S131–S153.
- Roh, D.S.; Du, Y.; Gabriele, M.L.; Robinson, A.R.; Niedernhofer, L.J.; Funderburgh, J.L. Age-related dystrophic changes in corneal endothelium from DNA repair-deficient mice. Aging Cell 2013, 12, 1122–1131.
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54.
- Roh, D.; Du, Y.; Robinson, A.; Gabriele, M.; Niedernhofer, L.; Funderburgh, J. Corneal Endothelial Changes in DNA Repair-Deficient Mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4290.
- Deshpande, N.; Melangath, G.; Vasanth, S.; Price, M.; Price, F.; Jurkunas, U.V. Defective DNA Repair in Fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 838.
- Deshpande, P.; Notara, M.; Bullett, N.; Daniels, J.T.; Haddow, D.B.; MacNeil, S. Development of a Surface-Modified Contact Lens for the Transfer of Cultured Limbal Epithelial Cells to the Cornea for Ocular Surface Diseases. Tissue Eng. Part A 2009, 15, 2889–2902.
- Asahina, H.; Han, Z.; Kawanishi, M.; Kato, T., Jr.; Ayaki, H.; Todo, T.; Yagi, T.; Takebe, H.; Ikenaga, M.; Kimura, S.H. Expression of a mammalian DNA photolyase confers light-dependent repair activity and reduces mutations of UV-irradiated shuttle vectors in xeroderma pigmentosum cells. Mutat. Res. 1999, 435, 255–262.
- Zwetsloot, J.C.; Vermeulen, W.; Hoeijmakers, J.H.; Yasui, A.; Eker, A.P.; Bootsma, D. Microinjected photoreactivating enzymes from Anacystis and Saccharomyces monomerize dimers in chromatin of human cells. Mutat. Res. 1985, 146, 71–77.
- Jans, J.; Schul, W.; Sert, Y.G.; Rijksen, Y.; Rebel, H.; Eker, A.P.; Nakajima, S.; van Steeg, H.; de Gruijl, F.R.; Yasui, A.; et al. Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol. 2005, 15, 105–115.
- Schul, W.; Jans, J.; Rijksen, Y.M.; Klemann, K.H.; Eker, A.P.; de Wit, J.; Nikaido, O.; Nakajima, S.; Yasui, A.; Hoeijmakers, J.H.; et al. Enhanced repair of cyclobutane pyrimidine dimers and improved UV resistance in photolyase transgenic mice. EMBO J. 2002, 21, 4719–4729.
- Zelle, B.; Reynolds, R.J.; Kottenhagen, M.J.; Schuite, A.; Lohman, P.H. The influence of the wavelength of ultraviolet radiation on survival, mutation induction and DNA repair in irradiated Chinese hamster cells. Mutat. Res. 1980, 72, 491–509.
- van Zeeland, A.A.; Smith, C.A.; Hanawalt, P.C. Sensitive determination of pyrimidine dimers in DNA of UV-irradiated mammalian cells. Introduction of T4 endonuclease V into frozen and thawed cells. Mutat. Res. 1981, 82, 173–189.
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369.
- Collins, A.R.; Mitchell, D.L.; Zunino, A.; de Wit, J.; Busch, D. UV-sensitive rodent mutant cell lines of complementation groups 6 and 8 differ phenotypically from their human counterparts. Environ. Mol. Mutagen. 1997, 29, 152–160.
- Boros, G.; Miko, E.; Muramatsu, H.; Weissman, D.; Emri, E.; Rozsa, D.; Nagy, G.; Juhasz, A.; Juhasz, I.; van der Horst, G.; et al. Transfection of pseudouridine-modified mRNA encoding CPD-photolyase leads to repair of DNA damage in human keratinocytes: A new approach with future therapeutic potential. J. Photochem. Photobiol. B 2013, 129, 93–99.
- Lopez-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Perez-Plasencia, C.; Alvarez-Sanchez, E.; Marchat, L.A. Protein Kinases and Transcription Factors Activation in Response to UV-Radiation of Skin: Implications for Carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172.
- Boros, G.; Miko, E.; Muramatsu, H.; Weissman, D.; Emri, E.; van der Horst, G.T.; Szegedi, A.; Horkay, I.; Emri, G.; Kariko, K.; et al. Identification of Cyclobutane Pyrimidine Dimer-Responsive Genes Using UVB-Irradiated Human Keratinocytes Transfected with In Vitro-Synthesized Photolyase mRNA. PLoS ONE 2015, 10, e0131141.
- Marizcurrena, J.J.; Martinez-Lopez, W.; Ma, H.; Lamparter, T.; Castro-Sowinski, S. A highly efficient and cost-effective recombinant production of a bacterial photolyase from the Antarctic isolate Hymenobacter sp. UV11. Extremophiles 2019, 23, 49–57.
- Yasuda, S.; Sekiguchi, M. T4 endonuclease involved in repair of DNA. Proc. Natl. Acad. Sci. USA 1970, 67, 1839–1845.

