Ultraviolet (UV) irradiation induces DNA lesions in all directly exposed tissues. In the human body, two tissues are chronically exposed to UV: the skin and the cornea. The most frequent UV-induced DNA lesions are cyclobutane pyrimidine dimers (CPDs) that can lead to apoptosis or induce tumorigenesis. Lacking the protective pigmentation of the skin, the transparent cornea is particularly dependent on nucleotide excision repair (NER) to remove UV-induced DNA lesions. The DNA damage response also triggers intracellular autophagy mechanisms to remove damaged material in the cornea. Therapeutic solutions involving xenogenic DNA-repair enzymes such as T4 endonuclease V or photolyases exist and are widely distributed for dermatological use.
1. Cornea
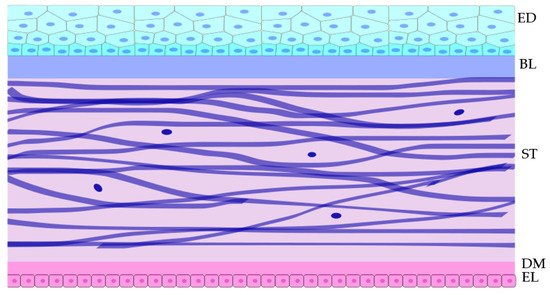
The cornea is the superficial shield at the front of the eye. Its transparency is essential for the transmission of light into the eye and through to the retina, enabling visual perception. The cornea functions as a physical barrier, protecting the inner contents of the eye, while also providing a significant portion of the refraction needed for proper vision. The human cornea consists of five distinct layers. The anterior layer is the epithelium, and its underlying fibrous mesh is called the Bowman layer [
1]. Posterior to the Bowman’s layer is the collagen-rich stroma, contributing to approximately 90% of the total corneal thickness. Beneath the stroma is Descemet’s membrane, which separates stroma from the most posterior corneal layer, the single cell layer endothelium (see
Figure 1) [
2,
3].
Figure 1. Schematic cross-section of the tissue layers within the central cornea and the approximate shape of cell within the layers. The top cyan layer is the epithelium,;below it is the acellular Bowman layer. Below that in violet are the fibroblasts within the stroma. Below the stroma is the acellular Descemet’s membrane to which the monolayer of pink endothelial cells adhere. Epithelial (ED), Bowman layer (BL), Stroma (ST), Descemet’s membrane (DM), and Endothelial layer (EL).
It is currently thought that the limbus and its inhabiting cells maintain the border between the two different tissues, and that any damage to the limbus may enable an invasion of conjunctival cells into the cornea. This typically leads to vascularization and a loss in vision quality [
10,
11,
12]. These disturbances may occur following UV damage where the limbal stem cells no longer maintain the border [
13,
14,
15,
16]. Although the role of epithelial factors in the maintenance of angiogenic and lymphangiogenic privilege seems evident, the role of the limbus as a physical barrier to vascular invasion seems poorly supported [
17,
18].
Damage to the epithelium must be repaired rapidly to restore barrier function and protect the cornea from bacteria or further trauma [
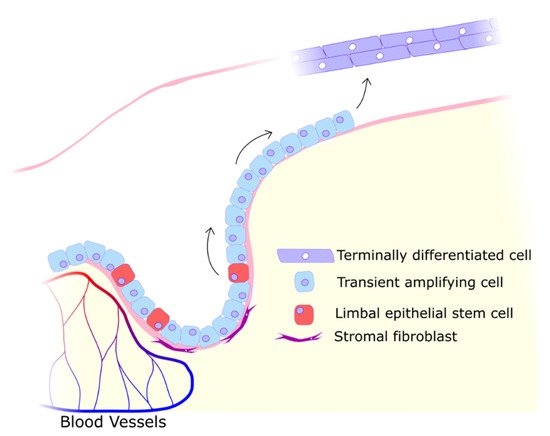
19]. As the epithelial cell population is maintained by limbal stem cells (see
Figure 2), the closure of wounds is severely impeded by any damage to their niche, the limbus [
20]. The limbus can be considered a particularly critical component of the cornea as this perimeter of crypts is essential in maintaining a clear cornea fully covered in epithelium. While the limbus is vulnerable to threats ranging from viral and bacterial to chemical and traumatic, one particular source of stress is almost always present: UV radiation.
Figure 2. Limbal stem cells differentiating into migratory transient amplifying cells that migrate along the Bowman layer into the avascular central cornea to proliferate and terminally differentiate into functional epithelial cells. The limbal niche is maintained by proximity to blood vessels and secreted factors from stromal cells.
2. UV Effects on the Cornea
2.1. UV Damage and Repair Mechanisms
In the case of the cornea, acute UV exposure is known to cause photokeratitis, a painful inflammation where the epithelium, stroma, and endothelium may be affected and which leads to clouding [26,27,28,29]. Chronic UV exposure usually leads to long term conditions such as tumours (squamous cell carcinomas, malignant melanomas, lymphoma of the conjunctiva [30,31,32]) or keratopathy [33,34,35].
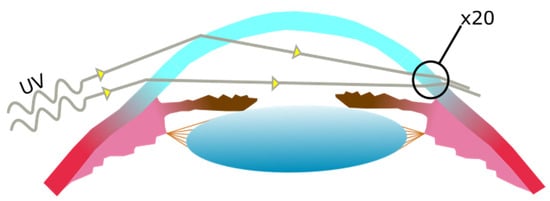
UV radiation entering the temporal limbus has been shown to focus at the nasal limbus, damaging limbal cells to a greater degree. This occurs specifically with UV entering at the temporal limbus that is then concentrated to the nasal limbus (see
Figure 3) [
36]. These damaged limbal cells and their descendants may then migrate from the limbus towards the center of the cornea [
37].
Figure 3. UV light entering the cornea laterally at the temporal side of the cornea, becoming focused at the nasal side of the cornea. The increase in UV exposure at the nasal side is estimated to be of up to twenty times the exposure on the temporal side.
2.2. Pterygium Aetiology and Pathogenesis
One of the first clear definitions of pterygium describes a pterygos, a wing-shaped degenerative and hyperplastic process where the bulbar conjunctiva invades onto the cornea [
66]. These vascularized, fibrotic degenerations continuously advance across the cornea over time [
67]. It is well documented across several ethnicities, locations, and age-groups that the primary risk factor of pterygia is UV [
68,
69,
70,
71,
72,
73]. Pterygia are typically found at the interpalpebral zone of the cornea. Due to the peripheral light focusing effect, they will develop more often at nasal side and less often on the temporal side of the cornea [
76].
2.3. UV-Induced DNA Lesion Formation
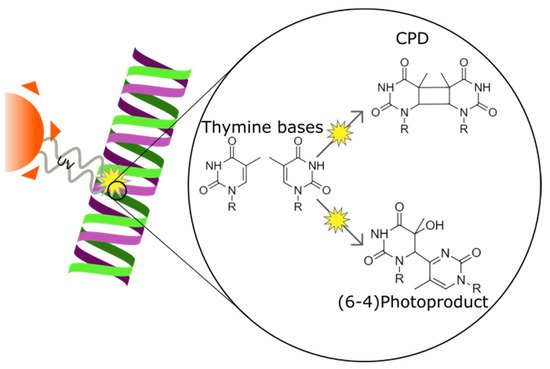
UV irradiation damages cellular DNA by causing the formation of cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs) (see
Figure 4) [
77]. CPD and 6-4PP lesions are both results of neighbouring pyrimidine covalently binding to each other [
78,
79,
80]. CPDs are the most frequent UV lesions and—when cells fail to induce cell death through the activation of the DNA damage response—lead to mutations that in turn promote malignancy [
81,
82,
83]. Mutations amid CPDs are responsible for transition mutations via Cytosine and Thymine nucleotide dimerization [
84,
85].
Figure 4. Ultraviolet radiation causing cyclobutane pirymidine dimer and pyrimidine-pyrimidone (6-4) photoproduct formation in a DNA strand between two thymine bases. The two forms of DNA damage differ in the position of the bond created between the two bases.
Photolyases exist in organisms ranging from bacteria to plants and marsupials but are absent in placental mammals. Humans in fact lack CPD and 6-4PP photolyases and instead rely on nuclear excision repair (NER) pathway [
92,
93]. NER is far more versatile in removing a range of helix distorting lesions; however, they are far less effective to recognize CPD and 6-4PP lesions than the photolyases that have been selected to bind to only those lesions with high specificity. In contrast to the single enzyme photolyases, NER is executed by a highly complex mechanism involving dozens of proteins. While these proteins will not necessarily target UV damage specifically like the aforementioned photolyases, they are capable of repairing CPD and 6-4PP [
94,
95]. NER is initiated by lesion recognition by either the Xeroderma pigmentosum complementation group C (XPC) acting in concert with the RAD23 homolog B (RAD23B) as part of global genome (GG-) NER or by the Cockayne syndrome B (CSB) protein encoded by the excision repair cross complementation group 6 (ERCC6) gene as part of transcription-coupled (TC-) NER, dependent on the location of the damage. TC-NER and GG-NER differ in the way they recognise DNA damage. GG-NER relies on the XPC complex constantly probing the DNA for lesions while TC-NER relies on RNA polymerase stalling at lesion sites to recognize damaged DNA [
96].
Both GG- and TC-NER recruit the NER core machinery including XPA and the TFIIH complex that unwind the double helix, verify the lesion, and subsequently recruit the ERCC1-XPF and the XPG endonucleases to excise a stretch containing the lesion [
97]. Mutations in NER genes typically result in UV hypersensitivity of the skin and particularly when GG-NER is affected in a several thousand fold higher incidence of skin cancer in Xeroderma pigmentosum (XP) patients. In contrast, mutations affecting TC-NER result in Cockayne syndrome (CS) that is characterized by growth retardation and premature aging but not skin cancer. Distinct mutations in the TFIIH components XPD can lead to XP, rare combinations of XP and CS as well as trichothiodystrophy (TTD) that shares many features with CS but in addition leads to transcription elongation defects that cause brittle hair and nails [
98,
99]. These three diseases can also present eye-related anomalies such as conjunctivitis, photophobia, keratitis, pterygium, and corneal opacity [
100], although these symptoms do not manifest in all patients.
2.4. The Role of Genotoxic Stress
The reason for the cornea’s greater resistance to UV lies in its ability to deal with genotoxic stress. Specifically, components of the genotoxic stress detection and response cascade such as DNA damage-binding protein 2 (DDB2), XPC, and tumour suppressor p53 are present in greater amounts in corneal epithelial cells than in epidermal keratinocyte [
108]. The quantity of a protein within a cell is a result of the balance between production and degradation. In the corneal epithelial cells, the degradation of XPC, DDB2, and p53 is slowed such that the active forms of these proteins are more common [
108]. While it seems like the cornea is better equipped than the skin to deal with UV damage, this could be interpreted as a compensation measure because the skin has corneocytes that form a physical barrier and prevent some genotoxic stress from occurring in the first place. The cornea instead relies on the tear film, which does not present as effective a barrier to UV [
109,
110].
2.5. Reactive Oxygen Species
UV may also damage epithelium by causing the formation of reactive oxygen species (ROS), free radicals that may oxidise cellular material such as proteins, organelles, or DNA [
118]. When the amount of ROS in a cell exceeds the cellular antioxidant levels, damage will occur more readily and the cells may undergo apoptosis [
119].
2.6. Disruption of Autophagy Mechanisms
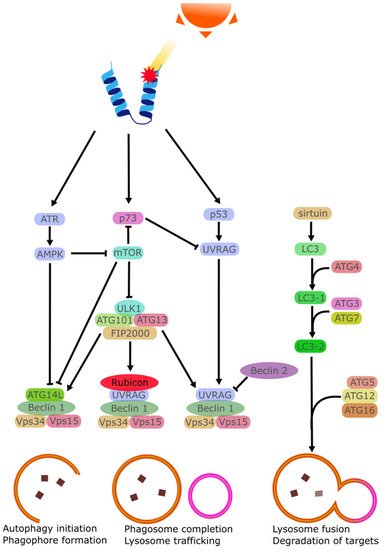
Autophagy is a degradation mechanism that utilises the lysosome to remove proteins from the cell. This is done either as part of the natural turnover of proteins or as part of a stress response. The process is mediated by several regulators that transport the targeted proteins and control the rate at which the proteins are removed. This rate can be adjusted by several stimuli to remove damaged proteins. One of these stimuli is UV irradiation. Ataxia-telangiectasia mutated (ATM) and Rad3-related (ATR) are both damage sensors that can mediate the activation of p53 via checkpoint kinase 1 (CHK1) and checkpoint kinase 2 (CHK2). ATR may also activate the STK11/AMPK metabolic pathway to stimulate the tuberous sclerosis 2 (TSC2) tumor suppressor and regulate autophagy via Beclin 1 [
130,
131]. TSC1 and TSC2 may also downregulate mTOR activity [
132]. The difference between ATM and ATR is that the former is an ATM-Chk2 pathway mostly activated by double-strand breaks, while the latter ATR/Chk1 is activated by single-strand breaks [
133,
134]. Both ATR and ATM inactivate mTORC1, enabling autophagy as a result [
135].
UV has also been found to stabilize p53, enabling it to be one of the starting points of the cellular response to UV stress by greatly increasing the amount of p53 within the cell [
136]. In enucleated cells, the inhibition of p53 leads to increased autophagy, indicating that the cytoplasmic p53 regulates autophagy [
137].
Autophagy is a continuous process with several regulating factors. Following the initial formation of the early phagosome, The LC3 will begin separately to finalize the phagosome formation and allow it to be trafficked to initiate the process of lysosomal breakdown. This process involves the Atg10/Atg7-mediated process of Atg5-Atg12-Atg16 conjugation [
146]. The sirtuin family has been documented as regulator of autophagy via this LC3 cascade [
147]. It should be noted that the seven members of the sirtuin family respond to different stimuli and may even inhibit autophagy given the right circumstances [
148].
While these autophagy mechanisms (see
Figure 5) are thoroughly researched, their role in corneal damage repair and particularly their activation following UV damage, is poorly characterised. Autophagy modulating treatments such as resveratrol eye drops are already being researched to address several eye diseases [
157,
158,
159,
160], but their exact effect on corneal autophagy in not known. It had already been acknowledged that autophagy likely plays an important role in some UV-induced and UV-propagated diseases such as dry-eye disease and pterygium [
161,
162].
Figure 5. Simplified schematic representation of the autophagic cascade activated by UV damage of proteins.
2.7. Apoptosis
Apoptosis can be triggered by several factors such as cytokines, hormones, virus, or drugs. At the right concentrations, these stimuli lead to the activation of effector caspases that trigger the apoptotic program [166]. UV irradiation has been shown to cause the activation of ATP-sensitive potassium (K(ATP)) channels in affected cells causing a loss of K+ ions and a depolarization of the mitochondrial membrane [167,168]. Intracellular K+ is importantly linked with the inhibition of caspase, and K+ loss is linked with greater rates of apoptosis [169]. In the cornea specifically, it has been observed that while in vivo cells did not perish following exposure to background doses of UVB, cultured cells did perish when exposed to similar UVB doses [170]. It was found that tear film provided a high amount of K+ ions to the corneal epithelia, ensuring a degree of apoptosis-resistance to the chronically UV-irradiated epithelial cells [171]. This is in contrast with the skin which lacks this excess K+ and instead regularly sees UV-triggered apoptosis in the form of sunburn cells [165,172].
3. UV Pathogenesis and Rescue
The failure of DNA repair pathways, particularly NER, typically results in developmental disorders, neurological symptoms, and skin cancers. These conditions arise due to the inability of cells to repair DNA in populations of progenitor cells, noncycling cells, and UV-exposed cells [
173]. While the disease itself is variable, one common trait unifies all of them: DNA photolesions are not being repaired.
The simplest method to prevent DNA photolesions is the avoidance of UV light, be it sunlight or artificial.
Early work on the failure of UV defense system performed in plants found that a loss of CPD photolyase activity resulted in hypersensitivity to UVB [
186]. CPD photolyase does not exist in placental mammals, [
187]. There exist animal models with DNA repair mutations that involve the other NER proteins that target CPD and 6-4PP. A model of systemic ERCC1 deficiency showed polyploidy in the liver and kidney [
188]. Polyploidy has been positively correlated to cell stress and senescence [
189]. A mouse model of ERCC1-knockdown in the corneal endothelium of saw decreased cell density in the corneal endothelium [
190]. Unfortunately, there are no reports of loss-of-function NER mutants specific to the corneal epithelia. Studies in humans looking at the NER expression profiles in Fuch’s endothelial corneal dystrophy (FECD) found that XPC was downregulated in patients that had FECD [
191,
192]. Although this does not make the cells more vulnerable to UV specifically, it does allow for faster accumulation of DNA lesions, which then lead to pathogenesis.
Because placental mammals lack CPD photolyase due to an evolutionary split, there has been some interest expressing CPD photolyase to potentially provide a much more competent repair enzyme. The photolyase in question is still expressed in marsupials and has been introduced into human cells harvested from an XP patient in an attempt to improve the rate of DNA repair. The marsupial photolyase functioned properly in the host cells and did improve survival of human cells following UV irradiation [
193]. Similar experiments that used microinjections of purified photolyases from yeast also found that UV-mediated cell death was reduced, but to a lesser degree compared to the marsupial photolyase [
194]. It was hypothesized that the yeast photolyase is not trafficked into the nucleus to the same degree as the marsupial photolyase. With such evidence, work continued with CPD photolyase introduction into placental mammals and a mouse line was generated that ubiquitously expressed either Potorous tridactylus CPD photolyase, Arabidopsis thaliana 6-4PP photolyase, or both [
195,
196]. The photoreactivation of CPD photolyase resulted in a markedly improved survival of UV-exposed cells in the mice, while photoreactivation of 6-4PPs did not noticeably affect UV-resistance. It should be noted that rodent cells have been documented multiple times as having CPD repair systems less effective than human CPD repair equivalents [
197,
198,
199,
200]. This lead to research on the introduction of CPD photolyase into mouse models and showed great improvement in recovery from UV damage [
195,
196].
Further work on repair of CPDs in human cells used an mRNA transfection system to express the marsupial Potorous tridactylus CPD photolyase in UVB-irradiated human epidermal keratinocytes. Results showed greatly reduced CPD amount and reduced activation of UV-induced cytokine such as IL-6 [
201]. Use of this system also lead to the identification of which genes where more highly expressed in the presence of CPDs, and thus could be targeted by therapeutic approaches [
87]. Cell cycle controllers like cyclin E1 and p15INK4b were expressed in greater quantities following JNK activation by UVB exposure. Several other genes were also identified with UV-dependent expression, although it is uncertain how beneficial the expression of each gene is [
202]. Work on the UV-induced CPD-dependent and CPD-independent cellular mechanisms continues, with CPD photolyase as a major tool. The obvious potential of CPD photolyase as a therapeutic agent in humans has been applied, usually with skin in mind [
203]. The potential of CPD photolyase in corneal healing comparatively sees less investigation, possibly due to the corneas greater success in dealing with CPDs with existing repair mechanisms or by simply changing the lifestyle of affected patients to better avoid UV exposure.
This entry is adapted from the peer-reviewed paper 10.3390/biology11020278