 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Teresa Cruz | + 2981 word(s) | 2981 | 2022-03-10 07:06:15 | | | |
| 2 | Peter Tang | Meta information modification | 2981 | 2022-03-18 12:18:36 | | |
Video Upload Options
Dendritic cells (DCs) are a class of bone-marrow-derived cells present in blood, epithelial, interstitial and lymphoid tissues, originated from lympho-myeloid hematopoiesis through a series of differentiation processes. Throughout the last decades, DC-based anti-tumor vaccines have proven to be a safe therapeutic approach, although with inconsistent clinical results. The functional limitations of ex vivo monocyte-derived dendritic cells (MoDCs) commonly used in these therapies are one of the pointed explanations for their lack of robustness. Among characterized human DC subpopulations, conventional type 1 DCs (cDC1) have emerged as a highly desirable tool for empowering anti-tumor immunity. This DC subset excels in its capacity to prime antigen-specific cytotoxic T cells and to activate natural killer (NK) and natural killer T (NKT) cells, which are critical factors for an effective anti-tumor immune response.
1. Introduction
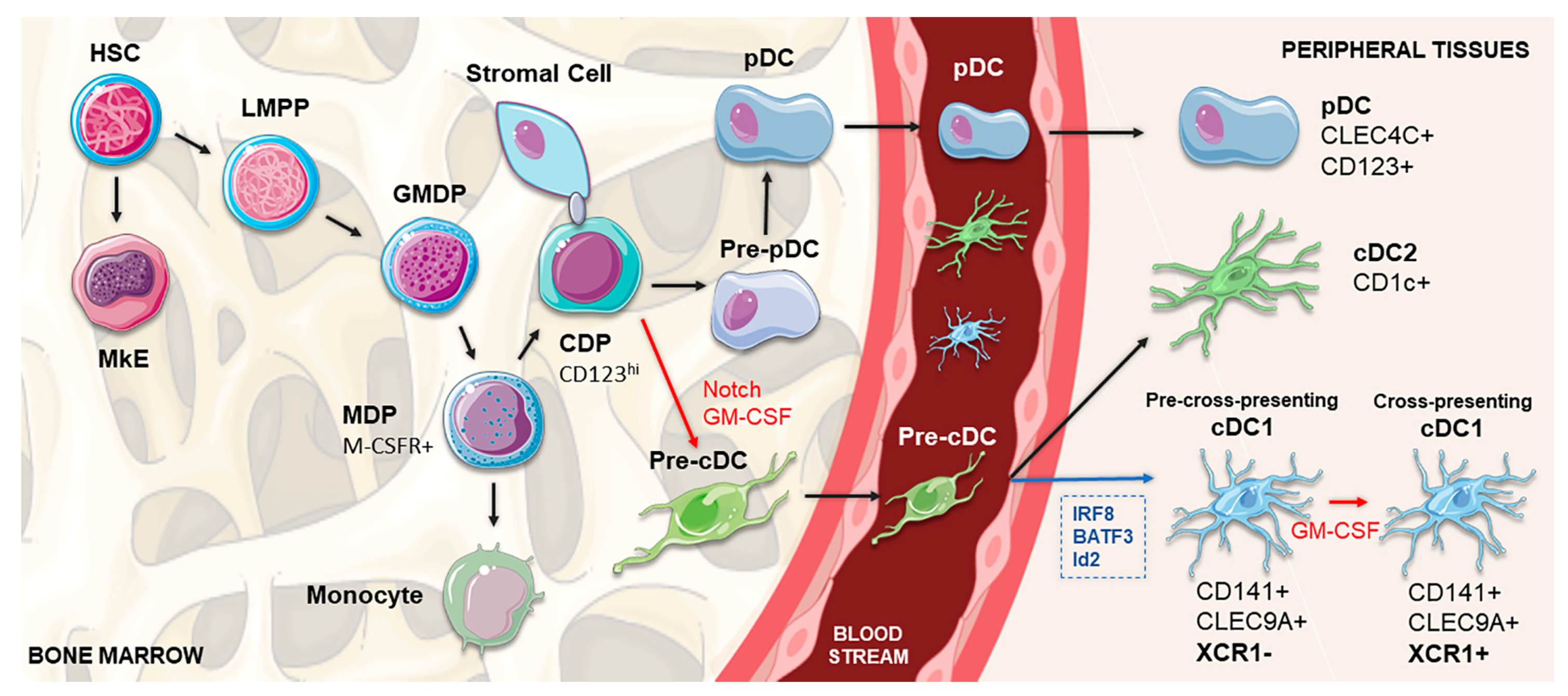
2. Development, Regulation and Heterogeneity of cDC1
3. The Role of cDC1 in Immunity

4. Exploiting cDC1 in Cancer Immunotherapy
|
Approach |
Studied Species |
Cell Subset |
Differentiation Cocktail |
Antigen Type |
Target/Tumor Model |
Combination Therapy |
Ref |
|---|---|---|---|---|---|---|---|
|
ex vivo differentiation |
Human |
CD34+-derived CD141+ CLEC9A+ DCs |
SCF, GM-CSF, IL-4 and Flt3L |
- |
- |
- |
|
|
Human |
CD34+-derived cDC1 |
Flt3L, SCF, TPO, IL-6 and StemRegenin1 |
- |
- |
- |
[79] |
|
|
Human |
Monocyte-derived CD141+ XCR1+ DCs 1 |
GM-CSF and IL-4 |
- |
- |
- |
[80] |
|
|
Human |
CD141+ XCR1+ DCs |
MA and LAM |
- |
- |
- |
[81] |
|
|
Human |
iPSC-derived CD141+ XCR1+ DCs |
GM-CSF, SCF, VEGF and BMP4 |
Melan A |
2 |
[82] |
||
|
Human and murine |
Fibroblast-derived cDC1 |
PU.1, IRF8 and BATF3 |
- |
- |
- |
[36] |
|
|
Naturally occurring cDC1 |
Murine |
Natural cDC1 |
- |
UV-irradiated tumor cell lysates |
B16 melanoma MC38 colon adenocarcinoma |
Anti-PD-1 |
[83] |
|
Murine |
Tumor-derived cDC1 |
- |
B16 melanoma LLC lung carcinoma |
[84] |
|||
|
mAb- or XCL1-based direct in vivo targeting |
Murine |
CD8α+ DC |
- |
IgG2a mAb Ovalbumin |
- |
- |
[85] |
|
Murine 3 |
XCR1+ DC |
- |
Ovalbumin |
EL4 thymoma |
- |
[86] |
|
|
Murine |
CD8α+ DC |
- |
Ovalbumin |
P3X63Ag8.653 myeloma |
- |
[87] |
|
|
Murine |
CD8α+ DC |
- |
Ovalbumin |
B16 melanoma |
- |
||
|
Murine |
CD8α+ DC |
- |
Ovalbumin |
B16 melanoma and lung pseudometastases |
- |
[90] |
|
|
Murine |
CD8α+ DC |
- |
MUC1 |
MC38 colon adenocarcinoma |
- |
[91] |
|
|
Murine |
CD8α+ DC |
- |
Nanoemulsion Ovalbumine |
PyMT-mChOVA breast cancer and lung metastases B16 melanoma HPV-related TC1 cancer |
- |
[92] |
|
|
Human 4 |
Allogeneic neuroblastoma cells |
- |
- |
Neuroblastoma |
- |
NCT01713439 NCT00703222 [93] |
|
|
Human 4 |
Autologous neuroblastoma cells |
- |
- |
Neuroblastoma |
- |
NCT00062855 [94] |
|
|
Human 4 |
Allogeneic neuroblastoma cells |
- |
- |
Neuroblastoma |
Cytoxan |
NCT01192555 |
|
|
WH-based direct in vivo targeting |
Murine |
CD8α+ DC |
- |
Ovalbumin |
B16 melanoma |
- |
[95] |
|
Indirect in vivo targeting |
Murine |
IFN- α -iPSC-pMCs |
B16 melanoma EL4 thymoma MC38 colon adenocarcinoma CT26 colorectal adenocarcinoma 4T1 breast cancer |
Anti-PD-1/anti-PD-L1 |
[96] |
||
|
Murine |
CD8α+ DC |
- |
Allogeneic T cells |
- |
- |
[97] |
1 Adherent fraction; 2 Functionality assays; 3 Transgenic mice expressing human XCR1; 4 Clinical trial.
References
- Oettgen, H.F. Immunotherapy of cancer. N. Engl. J. Med. 1977, 297, 484–491.
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322.
- Martin-Liberal, J.; Ochoa de Olza, M.; Hierro, C.; Gros, A.; Rodon, J.; Tabernero, J. The expanding role of immunotherapy. Cancer Treat. Rev. 2017, 54, 74–86.
- Adachi, K.; Tamada, K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015, 106, 945–950.
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461.
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267.
- Childs, R.W.; Carlsten, M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: The force awakens. Nat. Rev. Drug Discov. 2015, 14, 487–498.
- Manzo, T.; Heslop, H.E.; Rooney, C.M. Antigen-specific T cell therapies for cancer. Hum. Mol. Genet. 2015, 24, R67–R73.
- Campbell, A.M.; Decker, R.H. Mini-review of conventional and hypofractionated radiation therapy combined with immunotherapy for non-small cell lung cancer. Transl. Lung Cancer Res. 2017, 6, 220–229.
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor dendritic cell–based vaccines: Lessons from 20 years of clinical trials and future perspectives. Transl. Res. 2016, 168, 74–95.
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95.
- Okamoto, M.; Kobayashi, M.; Yonemitsu, Y.; Koido, S.; Homma, S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 133.
- Steinman, R.M. Decisions about Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2011, 30, 1–22.
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426.
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48.
- Calmeiro, J.; Carrascal, M.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Biomaterial-based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J. Immunother. Cancer 2019, 7, 238.
- Cintolo, J.A.; Datta, J.; Mathew, S.J.; Czerniecki, B.J. Dendritic cell-based vaccines: Barriers and opportunities. Future Oncol. 2012, 8, 1273–1299.
- Saxena, M.; Balan, S.; Roudko, V.; Bhardwaj, N. Towards superior dendritic-cell vaccines for cancer therapy. Nat. Biomed. Eng. 2018, 2, 341–346.
- Wimmers, F.; Schreibelt, G.; Sköld, A.E.; Figdor, C.G.; De Vries, I.J.M. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front. Immunol. 2014, 5, 165.
- Verdijk, P.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Boullart, A.C.I.; Kok, E.; van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.M.; et al. Limited Amounts of Dendritic Cells Migrate into the T-Cell Area of Lymph Nodes but Have High Immune Activating Potential in Melanoma Patients. Clin. Cancer Res. 2009, 15, 2531–2540.
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ Dendritic Cells Derived In Vitro from CD34+ Progenitors Closely Resemble Blood Dendritic Cells, Including Their Adjuvant Responsiveness, Contrary to Monocyte-Derived Dendritic Cells. J. Immunol. 2014, 193, 1622–1635.
- Romano, E.; Rossi, M.; Ratzinger, G.; de Cos, M.-A.; Chung, D.J.; Panageas, K.S.; Wolchock, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-Loaded Langerhans Cells, Despite Increased IL15 Secretion and T-Cell Activation In Vitro, Elicit Antitumor T-Cell Responses Comparable to Peptide-Loaded Monocyte-Derived Dendritic Cells In Vivo. Clin. Cancer Res. 2011, 17, 1984–1997.
- Ratzinger, G.; Baggers, J.; de Cos, M.A.; Yuan, J.; Dao, T.; Reagan, J.L.; Münz, C.; Heller, G.; Young, J.W. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monoc. J. Immunol. 2004, 173, 2780–2791.
- Tel, J.; Schreibelt, G.; Sittig, S.P.; Mathan, T.S.M.; Buschow, S.I.; Cruz, L.J.; Lambeck, A.J.A.; Figdor, C.G.; de Vries, I.J.M. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013, 121, 459–467.
- Haniffa, M.; Collin, M.; Ginhoux, F. Ontogeny and functional specialization of dendritic cells in human and mouse. Adv. Immunol. 2013, 120, 1–49.
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20.
- Takeuchi, S.; Furue, M. Dendritic cells: Ontogeny. Allergol. Int. 2007, 56, 215–223.
- Castell-Rodríguez, A.; Piñón-Zárate, G.; Herrera-Enríquez, M.; Jarquín-Yáñez, K.; Medina-Solares, I. Dendritic Cells: Location, Function, and Clinical Implications. In Biology of Myelomonocytic Cells; IntechOpen: London, UK, 2017.
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604.
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20.
- Dresch, C.; Leverrier, Y.; Marvel, J.; Shortman, K. Development of antigen cross-presentation capacity in dendritic cells. Trends Immunol. 2012, 33, 381–388.
- Breton, G.; Lee, J.; Zhou, Y.J.; Schreiber, J.J.; Keler, T.; Puhr, S.; Anandasabapathy, N.; Schlesinger, S.; Caskey, M.; Liu, K.; et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J. Exp. Med. 2015, 212, 401–413.
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.
- Bachem, A.; Hartung, E.; Guttler, S.; Mora, A.; Zhou, X.; Hegemann, A.; Plantinga, M.; Mazzini, E.; Stoitzner, P.; Gurka, S.; et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 2012, 3, 214.
- Miller, J.C.; Brown, B.D.; Shay, T.; Gautier, E.L.; Jojic, V.; Cohain, A.; Pandey, G.; Leboeuf, M.; Elpek, K.G.; Helft, J.; et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 2012, 13, 888–899.
- Rosa, F.F.; Pires, C.F.; Kurochkin, I.; Ferreira, A.G.; Gomes, A.M.; Palma, L.G.; Shaiv, K.; Solanas, L.; Azenha, C.; Papatsenko, D.; et al. Direct reprogramming of fibroblasts into antigen-presenting dendritic cells. Sci. Immunol. 2018, 3, 1–16.
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271.
- Balan, S.; Arnold-Schrauf, C.; Abbas, A.; Couespel, N.; Savoret, J.; Imperatore, F.; Villani, A.C.; Vu Manh, T.P.; Bhardwaj, N.; Dalod, M. Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep. 2018, 24, 1902–1915.
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141 + (BDCA-3) + dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260.
- Haniffa, M.; Collin, M.; Ginhoux, F. Identification of human tissue cross-presenting dendritic cells: A new target for cancer vaccines. Oncoimmunology 2013, 2, e23140.
- Yamazaki, C.; Sugiyama, M.; Ohta, T.; Hemmi, H.; Hamada, E.; Sasaki, I.; Fukuda, Y.; Yano, T.; Nobuoka, M.; Hirashima, T.; et al. Critical Roles of a Dendritic Cell Subset Expressing a Chemokine Receptor, XCR1. J. Immunol. 2013, 190, 6071–6082.
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281.
- Zhang, J.G.; Czabotar, P.E.; Policheni, A.N.; Caminschi, I.; Wan, S.S.; Kitsoulis, S.; Tullett, K.M.; Robin, A.Y.; Brammananth, R.; van Delft, M.F.; et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 2012, 36, 646–657.
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; e Sousa, C.R. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903.
- Isolation, T.; Equivalents, I.V. Chapter 5 the Isolation and Enrichment of Large Numbers of Highly Purifi ed Mouse Spleen Dendritic Cell Populations and Their. Methods Mol. Biol. 2018, 1423, 61–87.
- Crozat, K.; Guiton, R.; Contreras, V.; Feuillet, V.; Dutertre, C.A.; Ventre, E.; Manh, T.P.V.; Baranek, T.; Storset, A.K.; Marvel, J.; et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1283–1292.
- Kroczek, R.A.; Henn, V. The Role of XCR1 and its Ligand XCL1 in Antigen Cross-Presentation by Murine and Human Dendritic Cells. Front. Immunol. 2012, 3, 14.
- Dorner, B.G.; Dorner, M.B.; Zhou, X.; Opitz, C.; Mora, A.; Güttler, S.; Hutloff, A.; Mages, H.W.; Ranke, K.; Schaefer, M.; et al. Selective Expression of the Chemokine Receptor XCR1 on Cross-presenting Dendritic Cells Determines Cooperation with CD8+ T Cells. Immunity 2009, 31, 823–833.
- Ohta, T.; Sugiyama, M.; Hemmi, H.; Yamazaki, C.; Okura, S.; Sasaki, I.; Fukuda, Y.; Orimo, T.; Ishii, K.J.; Hoshino, K.; et al. Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Sci. Rep. 2016, 6, 23505.
- Brewitz, A.; Eickhoff, S.; Dähling, S.; Quast, T.; Bedoui, S.; Kroczek, R.A.; Kurts, C.; Garbi, N.; Barchet, W.; Iannacone, M.; et al. CD8+ T Cells Orchestrate pDC-XCR1+ Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity 2017, 46, 205–219.
- Alexandre, Y.O.; Ghilas, S.; Sanchez, C.; Le Bon, A.; Crozat, K.; Dalod, M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. J. Exp. Med. 2016, 213, 75–92.
- Cancel, J.-C.; Crozat, K.; Dalod, M.; Mattiuz, R. Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front. Immunol. 2019, 10, 9.
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100.
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652.
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938.
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79.
- Wylie, B.; Seppanen, E.; Xiao, K.; Zemek, R.; Zanker, D.; Prato, S.; Foley, B.; Hart, P.H.; Kroczek, R.A.; Chen, W.; et al. Cross-presentation of cutaneous melanoma antigen by migratory XCR1 + CD103 − and XCR1 + CD103 + dendritic cells. Oncoimmunology 2015, 4, e1019198.
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103 + /CD141 + Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336.
- Michea, P.; Noël, F.; Zakine, E.; Czerwinska, U.; Sirven, P.; Abouzid, O.; Goudot, C.; Scholer-Dahirel, A.; Vincent-Salomon, A.; Reyal, F.; et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat. Immunol. 2018, 19, 885–897.
- Sluijter, B.J.R.; Van Den Hout, M.F.C.M.; Koster, B.D.; Van Leeuwen, P.A.M.; Schneiders, F.L.; Van De Ven, R.; Molenkamp, B.G.; Vosslamber, S.; Verweij, C.L.; Van Den Tol, M.P.; et al. Arming the melanoma sentinel lymph node through local administration of CpG-B and GM-CSF: Recruitment and activation of BDCA3/CD141+ dendritic cells and enhanced cross-presentation. Cancer Immunol. Res. 2015, 3, 495–505.
- Böttcher, J.P.; e Sousa, C.R. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792.
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458.
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural Killer Cell Accumulation in Tumors Is Dependent on IFN- and CXCR3 Ligands. Cancer Res. 2008, 68, 8437–8445.
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103+ Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108.
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637.
- Ferlazzo, G.; Morandi, B. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front. Immunol. 2014, 5, 159.
- Deauvieau, F.; Ollion, V.; Doffin, A.-C.; Achard, C.; Fonteneau, J.-F.; Verronese, E.; Durand, I.; Ghittoni, R.; Marvel, J.; Dezutter-Dambuyant, C.; et al. Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int. J. Cancer 2015, 136, 1085–1094.
- Wong, J.L.; Berk, E.; Edwards, R.P.; Kalinski, P. IL-18-Primed Helper NK Cells Collaborate with Dendritic Cells to Promote Recruitment of Effector CD8+ T Cells to the Tumor Microenvironment. Cancer Res. 2013, 73, 4653–4662.
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191.
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235.
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109.
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.J.G.; Duiveman-de Boer, T.; van de Rakt, M.W.M.M.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin. Cancer Res. 2016, 22, 2155–2166.
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302.
- Tel, J.; Aarntzen, E.H.J.G.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.G.; van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res. 2013, 73, 1063–1075.
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A Phase I Clinical Trial of CD1c (BDCA-1)+ Dendritic Cells Pulsed With HLA-A*0201 Peptides for Immunotherapy of Metastatic Hormone Refractory Prostate Cancer. J. Immunother. 2015, 38, 71–76.
- Balan, S.; Dalod, M. In vitro generation of human XCR1+ dendritic cells from CD34+ hematopoietic progenitors. Methods Mol. Biol. 2016, 1423, 19–37.
- Thordardottir, S.; Hangalapura, B.N.; Hutten, T.; Cossu, M.; Spanholtz, J.; Schaap, N.; Radstake, T.R.D.J.; Van Der Voort, R.; Dolstra, H. The aryl hydrocarbon receptor antagonist StemRegenin 1 promotes human plasmacytoid and myeloid dendritic cell development from CD34+ hematopoietic progenitor cells. Stem Cells Dev. 2014, 23, 955–967.
- Kim, S.J.; Kim, G.; Kim, N.; Chu, H.; Park, B.-C.; Yang, J.S.; Han, S.H.; Yun, C.-H. Human CD141+ dendritic cells generated from adult peripheral blood monocytes. Cytotherapy 2019, 21, 1049–1063.
- Tomita, Y.; Watanabe, E.; Shimizu, M.; Negishi, Y.; Kondo, Y.; Takahashi, H. Induction of tumor-specific CD8+ cytotoxic T lymphocytes from naïve human T cells by using Mycobacterium-derived mycolic acid and lipoarabinomannan-stimulated dendritic cells. Cancer Immunol. Immunother. 2019, 68, 1605–1619.
- Silk, K.M.; Silk, J.D.; Ichiryu, N.; Davies, T.J.; Nolan, K.F.; Leishman, A.J.; Carpenter, L.; Watt, S.M.; Cerundolo, V.; Fairchild, P.J. Cross-presentation of tumour antigens by human induced pluripotent stem cell-derived CD141+XCR1+ dendritic cells. Gene Ther. 2012, 19, 1035–1040.
- Wculek, S.K.; Amores-Iniesta, J.; Conde-Garrosa, R.; Khouili, S.C.; Melero, I.; Sancho, D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J. Immunother. Cancer 2019, 7, 1–16.
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 1–17.
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.Y.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.H.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273.
- Hartung, E.; Becker, M.; Bachem, A.; Reeg, N.; Jäkel, A.; Hutloff, A.; Weber, H.; Weise, C.; Giesecke, C.; Henn, V.; et al. Induction of Potent CD8 T Cell Cytotoxicity by Specific Targeting of Antigen to Cross-Presenting Dendritic Cells In Vivo via Murine or Human XCR1. J. Immunol. 2015, 194, 1069–1079.
- Kroczek, A.L.; Hartung, E.; Gurka, S.; Becker, M.; Reeg, N.; Mages, H.W.; Voigt, S.; Freund, C.; Kroczek, R.A. Structure-Function Relationship of XCL1 Used for in vivo Targeting of Antigen Into XCR1+ Dendritic Cells. Front. Immunol. 2018, 9, 2806.
- Mizumoto, Y.; Katsuda, M.; Miyazawa, M.; Kitahata, Y.; Miyamoto, A.; Nakamori, M.; Ojima, T.; Matsuda, K.; Hemmi, H.; Tamada, K.; et al. In Vivo Antigen Delivery to Dendritic Cells-A Novel Peptide Vaccine for Cancer Therapy. Cancer Chemother. 2018, 45, 1469–1471.
- Terhorst, D.; Fossum, E.; Baranska, A.; Tamoutounour, S.; Malosse, C.; Garbani, M.; Braun, R.; Lechat, E.; Crameri, R.; Bogen, B.; et al. Laser-Assisted Intradermal Delivery of Adjuvant-Free Vaccines Targeting XCR1 + Dendritic Cells Induces Potent Antitumoral Responses. J. Immunol. 2015, 194, 5895–5902.
- Sancho, D.; Mourão-sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110.
- Picco, G.; Beatson, R.; Taylor-Papadimitriou, J.; Burchell, J.M. Targeting DNGR-1 (CLEC9A) with antibody/MUC1 peptide conjugates as a vaccine for carcinomas. Eur. J. Immunol. 2014, 44, 1947–1955.
- Zeng, B.; Middelberg, A.P.J.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984.
- Rousseau, R.F.; Haight, A.E.; Hirschmann-Jax, C.; Yvon, E.S.; Rill, D.R.; Mei, Z.; Smith, S.C.; Inman, S.; Cooper, K.; Alcoser, P.; et al. Local and systemic effects of an allogeneic tumor cell vaccine combining transgenic human lymphotactin with interleukin-2 in patients with advanced or refractory neuroblastoma. Blood 2003, 101, 1718–1726.
- Russell, H.V.; Strother, D.; Mei, Z.; Rill, D.; Popek, E.; Biagi, E.; Yvon, E.; Brenner, M.; Rousseau, R. Phase I trial of vaccination with autologous neuroblastoma tumor cells genetically modified to secrete IL-2 and lymphotactin. J. Immunother. 2007, 30, 227–233.
- Yan, Z.; Wu, Y.; Du, J.; Li, G.; Wang, S.; Cao, W.; Zhou, X.; Wu, C.; Zhang, D.; Jing, X.; et al. A novel peptide targeting Clec9a on dendritic cell for cancer immunotherapy. Oncotarget 2016, 7, 40437–40450.
- Tsuchiya, N.; Zhang, R.; Iwama, T.; Ueda, N.; Liu, T.; Tatsumi, M.; Sasaki, Y.; Shimoda, R.; Osako, Y.; Sawada, Y.; et al. Type I Interferon Delivery by iPSC-Derived Myeloid Cells Elicits Antitumor Immunity via XCR1+ Dendritic Cells. Cell Rep. 2019, 29, 162–175.
- Kitazawa, Y.; Ueta, H.; Sawanobori, Y.; Katakai, T.; Yoneyama, H.; Ueha, S.; Matsushima, K.; Tokuda, N.; Matsuno, K. Novel targeting to XCR1+ dendritic cells using allogeneic T cells for polytopical antibody responses in the lymph nodes. Front. Immunol. 2019, 10, 1195.

