 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jun-Ping Liu | + 2538 word(s) | 2538 | 2022-03-07 09:46:09 | | | |
| 2 | Yvaine Wei | Meta information modification | 2538 | 2022-03-18 07:10:03 | | |
Video Upload Options
Pulmonary senescence is accelerated by unresolved DNA damage response, underpinning susceptibility to pulmonary fibrosis. Recently it was reported that the SARS-Cov-2 viral infection induces acute pulmonary epithelial senescence followed by fibrosis. Notably, the TGF-β signalling pathway mediates alveolar epithelial stem cell senescence by mechanisms involving suppression of the telomerase reverse transcriptase gene in pulmonary fibrosis. Alternatively, telomere uncapping caused by stress-induced telomeric shelterin protein TPP1 degradation mediates DNA damage response, pulmonary senescence and fibrosis. However, targeted intervention of cellular senescence disrupts pulmonary remodelling and fibrosis by clearing senescent cells using senolytics or preventing senescence using telomere dysfunction inhibitor (TELODIN). Studies indicate that the development of senescence-associated differentiation disorders is reprogrammable and reversible by inhibiting stem cell replicative senescence in pulmonary fibrosis, providing a framework for targeted intervention of the molecular mechanisms of alveolar stem cell senescence and pulmonary fibrosis.
1. Introduction
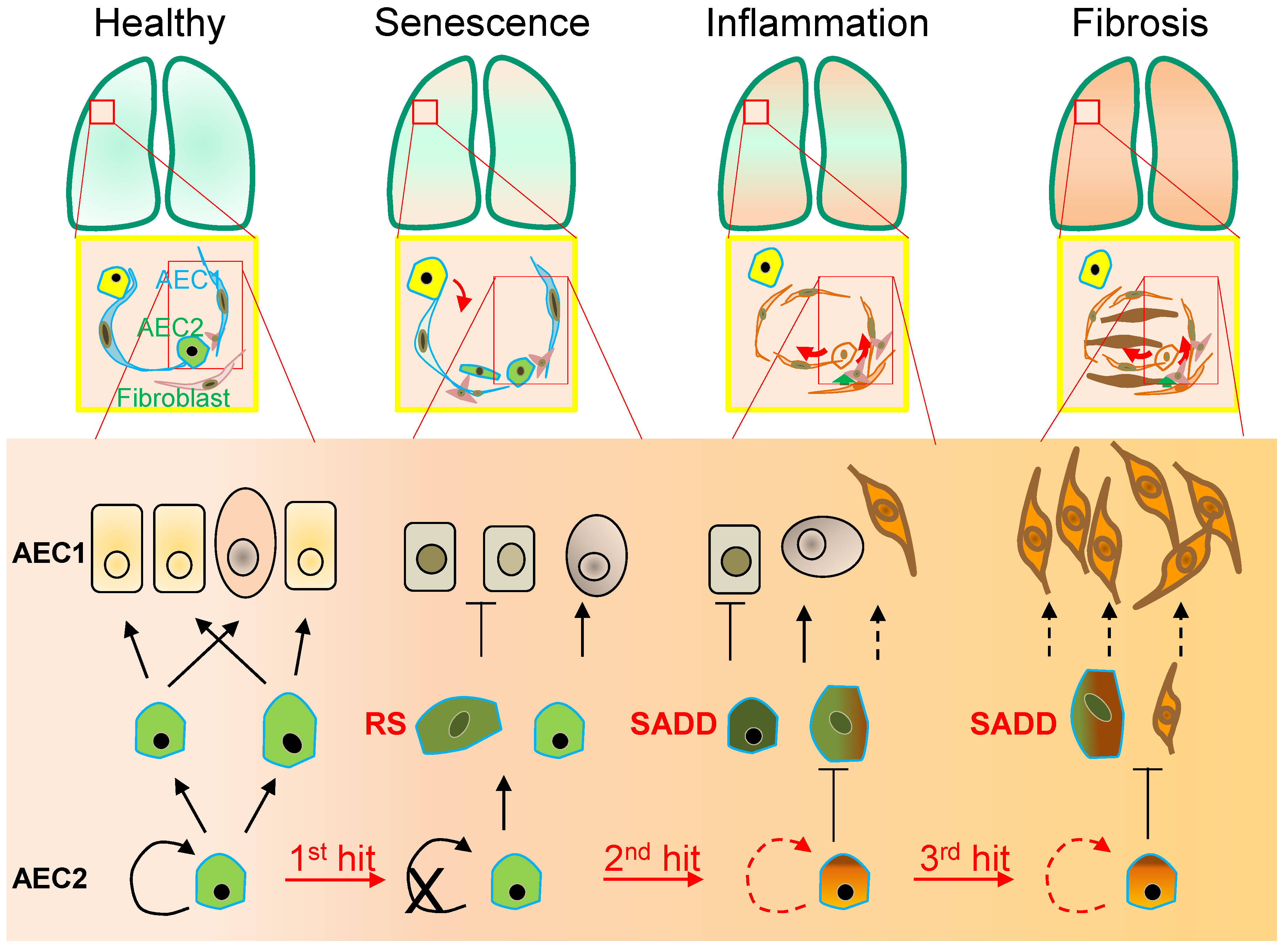
2. Senescence-Associated Differentiation Disorder (SADD)
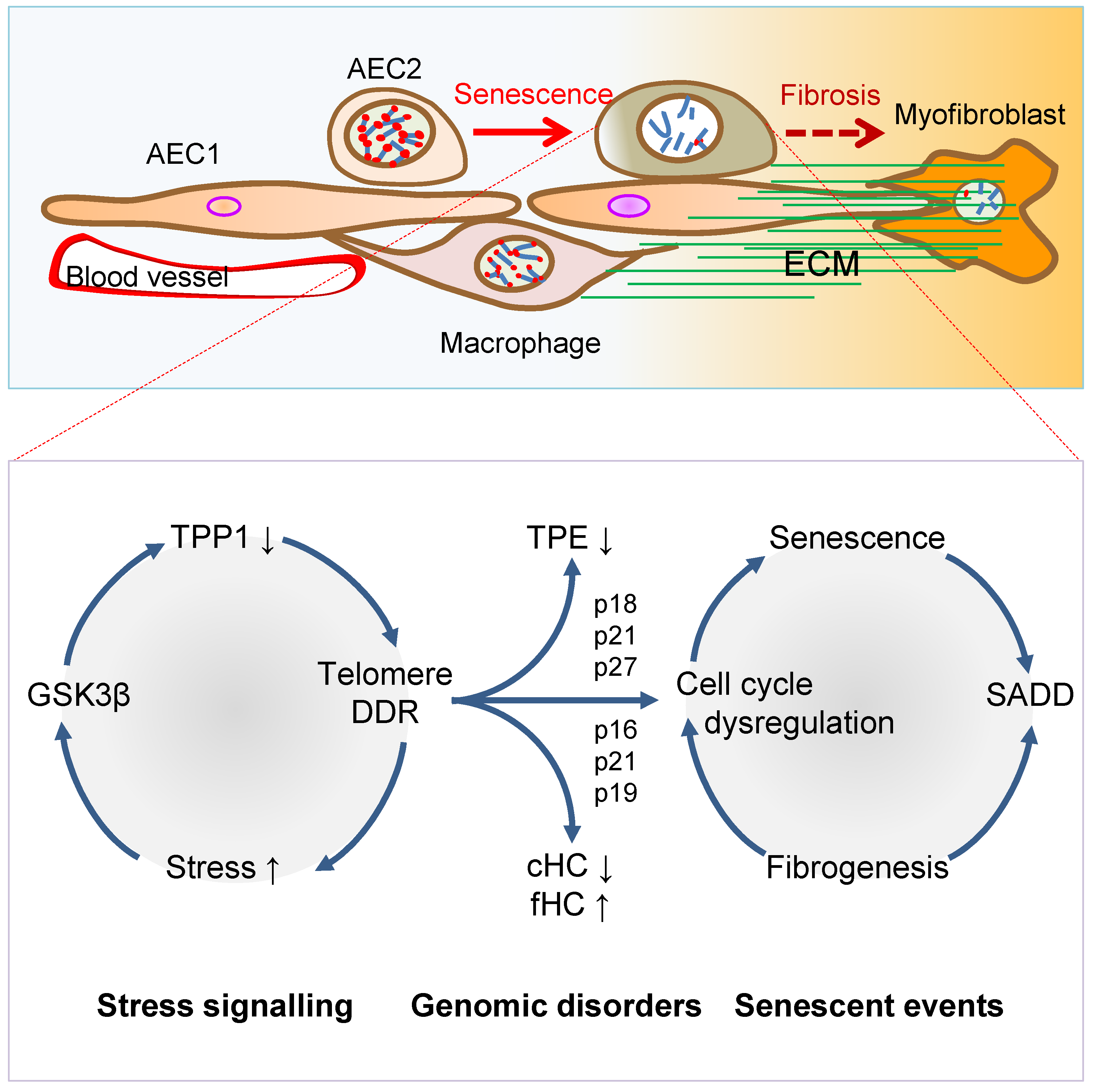
3. Telomere Dysfunction Mediates Pulmonary Senescence and Fibrosis
3.1. Telomerase Gene Deficiency in Ageing-Related Disorders
3.2. TGF-β Signaling to Telomerase TERT Gene Repression
3.3. Stress-Induced TPP1 Degradation and Telomere Uncapping
4. Targeted Intervention of Cellular Senescence and Tissue Fibrosis
4.1. Targeting Anti-Apoptotic Gene Bcl-2 to Clear Senescent Cells
4.2. Targeting TP53 and p16INK4A Tumor Suppressor Genes to Clear Senescent Cells
4.3. Preventing Telomere Dysfunction and Pulmonary Fibrosis by TELODIN
5. Conclusions
References
- Cookson, W.O.; Moffatt, M.F. Bedside to gene and back in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2013, 368, 2228–2230.
- Armanios, M.Y. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1370–1372.
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289.
- Wang, S.; Yao, X.; Ma, S.; Ping, Y.; Fan, Y.; Sun, S.; He, Z.; Shi, Y.; Sun, L.; Xiao, S.; et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 2021, 23, 1314–1328.
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 2021, 184, 775–791.e14.
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326.
- Ting, C.; Aspal, M.; Vaishampayan, N.; Huang, S.K.; Wang, F.; Farver, C.; Zemans, R.L. Ineffectual AEC1 differentiation from KRT8 (hi) transitional state without fibrosis is associated with fatal COVID-19 ARDS. bioRxiv 2021.
- Drake, T.M.; Docherty, A.B.; Harrison, E.M.; Quint, J.K.; Adamali, H.; Agnew, S.; Babu, S.; Barber, C.M.; Barratt, S.; Bendstrup, E.; et al. Outcome of Hospitalization for COVID-19 in Patients with Interstitial Lung Disease. An International Multicenter Study. Am. J. Respir. Crit. Care Med. 2020, 202, 1656–1665.
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 519–526.
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036.
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358.e4.
- Rao, W.; Wang, S.; Duleba, M.; Niroula, S.; Goller, K.; Xie, J.; Mahalingam, R.; Neupane, R.; Liew, A.A.; Vincent, M.; et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 2020, 181, 848–864.e18.
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014, 156, 440–455.
- Chen, Q.; Suresh Kumar, V.; Finn, J.; Jiang, D.; Liang, J.; Zhao, Y.Y.; Liu, Y. CD44(high) alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L41–L51.
- Reddy, R.; Buckley, S.; Doerken, M.; Barsky, L.; Weinberg, K.; Anderson, K.D.; Warburton, D.; Driscoll, B. Isolation of a putative progenitor subpopulation of alveolar epithelial type 2 cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, 658–667.
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la O, S.M.; Flynn, R.A.; Margalef-Catala, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675.
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123.
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7.
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946.
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559.
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8(hi) Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447.
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single cell RNA sequencing identifies TGFbeta as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019, 4, e123637.
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e4.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol 2014, 15, 482–496.
- Maciel-Baron, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martinez, F.; Galvan-Arzate, S.; Luna-Lopez, A.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Konigsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 2016, 38, 26.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495.
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017, 24, 30–39.
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466.
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538.e9.
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjo, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59.
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061.
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737.
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y.; Liu, J.P. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.e9.
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 5024–5033.
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717.
- Reyes, J.; Chen, J.Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591.e5.
- Baryshev, M.; Inashkina, I.; Salmina, K.; Huna, A.; Jackson, T.R.; Erenpreisa, J. DNA methylation of the Oct4A enhancers in embryonal carcinoma cells after etoposide treatment is associated with alternative splicing and altered pluripotency in reversibly senescent cells. Cell Cycle 2018, 17, 362–366.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100.
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming In Vivo. Science 2016, 354, aaf4445.
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183.
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176.
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522.
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. CB 2017, 27, 2652–2660.e4.
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729.
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287.
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871.
- Konopka, K.E.; Nguyen, T.; Jentzen, J.M.; Rayes, O.; Schmidt, C.J.; Wilson, A.M.; Farver, C.F.; Myers, J.L. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology 2020, 77, 570–578.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140.
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983.
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972.
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104.
- Razdan, N.; Vasilopoulos, T.; Herbig, U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 2018, 17, e12838.
- Lee, J.; Reddy, R.; Barsky, L.; Scholes, J.; Chen, H.; Shi, W.; Driscoll, B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, 57–70.
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332.
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A.; et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, 71–78.
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops In Vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185.
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224.
- Namba, T.; Tanaka, K.I.; Ito, Y.; Hoshino, T.; Matoyama, M.; Yamakawa, N.; Isohama, Y.; Azuma, A.; Mizushima, T. Induction of EMT-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell Death Differ. 2010, 17, 1882–1895.
- Schneider, D.J.; Wu, M.; Le, T.T.; Cho, S.H.; Brenner, M.B.; Blackburn, M.R.; Agarwal, S.K. Cadherin-11 contributes to pulmonary fibrosis: Potential role in TGF-beta production and epithelial to mesenchymal transition. FASEB J. 2012, 26, 503–512.
- Tanjore, H.; Cheng, D.S.; Degryse, A.L.; Zoz, D.F.; Abdolrasulnia, R.; Lawson, W.E.; Blackwell, T.S. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 2011, 286, 30972–30980.
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665.
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038.
- Zhang, K.; Wang, L.; Hong, X.; Chen, H.; Shi, Y.; Liu, Y.; Liu, J.; Liu, J.P. Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice. Cells 2021, 10, 2892.
- Chen, R.; Zhang, K.; Chen, H.; Zhao, X.; Wang, J.; Li, L.; Cong, Y.; Ju, Z.; Xu, D.; Williams, B.R.; et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 2015, 290, 30813–30829.
- Wang, L.; Yu, X.; Liu, J.P. Telomere Damage Response and Low-Grade Inflammation. Adv. Exp. Med. Biol 2017, 1024, 213–224.
- Birnhuber, A.; Egemnazarov, B.; Biasin, V.; Bonyadi Rad, E.; Wygrecka, M.; Olschewski, H.; Kwapiszewska, G.; Marsh, L.M. CDK4/6 inhibition enhances pulmonary inflammatory infiltration in bleomycin-induced lung fibrosis. Respir. Res. 2020, 21, 167.
- Sala, M.A.; Balderas-Martinez, Y.I.; Buendia-Roldan, I.; Abdala-Valencia, H.; Nam, K.; Jain, M.; Bhorade, S.; Bharat, A.; Reyfman, P.A.; Ridge, K.M.; et al. Inflammatory pathways are upregulated in the nasal epithelium in patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 233.
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287.
- Aumiller, V.; Balsara, N.; Wilhelm, J.; Gunther, A.; Konigshoff, M. WNT/beta-catenin signaling induces IL-1beta expression by alveolar epithelial cells in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 96–104.
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49.
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040.
- Otoupalova, E.; Smith, S.; Cheng, G.; Thannickal, V.J. Oxidative Stress in Pulmonary Fibrosis. Compr. Physiol. 2020, 10, 509–547.
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Kazemier, K.M.; Ruven, H.J.; Grutters, J.C. Association between variations in cell cycle genes and idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e30442.
- Dressen, A.; Abbas, A.R.; Cabanski, C.; Reeder, J.; Ramalingam, T.R.; Neighbors, M.; Bhangale, T.R.; Brauer, M.J.; Hunkapiller, J.; Reeder, J.; et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. Lancet Respir. Med. 2018, 6, 603–614.
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961.
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557.
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517.
- Sousa, S.R.; Caetano Mota, P.; Melo, N.; Bastos, H.N.; Padrao, E.; Pereira, J.M.; Cunha, R.; Souto Moura, C.; Guimaraes, S.; Morais, A. Heterozygous TERT gene mutation associated with familial idiopathic pulmonary fibrosis. Respir. Med. Case Rep. 2019, 26, 118–122.
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056.
- Alder, J.K.; Cogan, J.D.; Brown, A.F.; Anderson, C.J.; Lawson, W.E.; Lansdorp, P.M.; Phillips, J.A., 3rd; Loyd, J.E.; Chen, J.J.; Armanios, M. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011, 7, e1001352.
- Armanios, M. Syndromes of Telomere Shortening. Annu Rev. Genom. Hum. Genet. 2009, 62, 6405–6409.
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93.
- Chilosi, M.; Poletti, V.; Rossi, A. The pathogenesis of COPD and IPF: Distinct horns of the same devil? Respir. Res. 2012, 13, 3.
- Nogee, L.M.; Dunbar, A.E., 3rd; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579.
- Cottin, V.; Reix, P.; Khouatra, C.; Thivolet-Bejui, F.; Feldmann, D.; Cordier, J.F. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 2011, 66, 918–919.
- Nathan, N.; Giraud, V.; Picard, C.; Nunes, H.; Dastot-Le Moal, F.; Copin, B.; Galeron, L.; De Ligniville, A.; Kuziner, N.; Reynaud-Gaubert, M.; et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 2016, 25, 1457–1467.
- Wu, J.; McKeague, M.; Sturla, S.J. Nucleotide-Resolution Genome-Wide Mapping of Oxidative DNA Damage by Click-Code-Seq. J. Am. Chem Soc. 2018, 140, 9783–9787.
- Qian, W.; Kumar, N.; Roginskaya, V.; Fouquerel, E.; Opresko, P.L.; Shiva, S.; Watkins, S.C.; Kolodieznyi, D.; Bruchez, M.P.; Van Houten, B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 18435–18444.
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708.
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A., 3rd; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134.
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365.
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963.
- Aitken, M.L.; Dugowson, C.; Schmidt, R.A.; Fer, M. Bleomycin-induced pulmonary fibrosis in a patient with rheumatoid arthritis. A possible synergistic effect? West. J. Med. 1989, 150, 344–346.
- Santrach, P.J.; Askin, F.B.; Wells, R.J.; Azizkhan, R.G.; Merten, D.F. Nodular form of bleomycin-related pulmonary injury in patients with osteogenic sarcoma. Cancer 1989, 64, 806–811.
- Parfrey, H.; Babar, J.; Fiddler, C.A.; Chilvers, E.R. Idiopathic pulmonary fibrosis in a Christmas Island nuclear test veteran. BMJ Case Rep. 2010, 2010, bcr0620103102.
- Desai, M.Y.; Karunakaravel, K.; Wu, W.; Agarwal, S.; Smedira, N.G.; Lytle, B.W.; Griffin, B.P. Pulmonary fibrosis on multidetector computed tomography and mortality in patients with radiation-associated cardiac disease undergoing cardiac surgery. J. Thorac. Cardiovasc. Surg. 2014, 148, 475–481.e3.
- Gross, N.J. Pulmonary effects of radiation therapy. Ann. Intern. Med. 1977, 86, 81–92.
- Chung, K.P.; Hsu, C.L.; Fan, L.C.; Huang, Z.; Bhatia, D.; Chen, Y.J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390.
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135.
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242.
- Borok, Z.; Buhl, R.; Grimes, G.J.; Bokser, A.D.; Hubbard, R.C.; Holroyd, K.J.; Roum, J.H.; Czerski, D.B.; Cantin, A.M.; Crystal, R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet 1991, 338, 215–216.
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198.
- Aalbers, A.M.; Kajigaya, S.; van den Heuvel-Eibrink, M.M.; van der Velden, V.H.; Calado, R.T.; Young, N.S. Human telomere disease due to disruption of the CCAAT box of the TERC promoter. Blood 2012, 119, 3060–3063.
- Hao, L.Y.; Armanios, M.; Strong, M.A.; Karim, B.; Feldser, D.M.; Huso, D.; Greider, C.W. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005, 123, 1121–1131.
- Fogarty, P.F.; Yamaguchi, H.; Wiestner, A.; Baerlocher, G.M.; Sloand, E.; Zeng, W.S.; Read, E.J.; Lansdorp, P.M.; Young, N.S. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 2003, 362, 1628–1630.
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395.
- Mitchell, J.R.; Wood, E.; Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402, 551–555.
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299.
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337.
- Blackburn, E.H.; Greider, C.W.; Henderson, E.; Lee, M.S.; Shampay, J.; Shippen-Lentz, D. Recognition and elongation of telomeres by telomerase. Genome 1989, 31, 553–560.
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352.
- Harle-Bachor, C.; Boukamp, P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 6476–6481.
- Counter, C.M.; Hahn, W.C.; Wei, W.; Caddle, S.D.; Beijersbergen, R.L.; Lansdorp, P.M.; Sedivy, J.M.; Weinberg, R.A. Dissociation among In Vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA 1998, 95, 14723–14728.
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574.
- Collins, K. Mammalian telomeres and telomerase. Curr. Opin. Cell Biol. 2000, 12, 378–383.
- Mitchell, J.R.; Collins, K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol. Cell 2000, 6, 361–371.
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235.
- Maida, Y.; Yasukawa, M.; Masutomi, K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2016, 36, 1248–1259.
- Yasukawa, M.; Ando, Y.; Yamashita, T.; Matsuda, Y.; Shoji, S.; Morioka, M.S.; Kawaji, H.; Shiozawa, K.; Machitani, M.; Abe, T.; et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat. Commun. 2020, 11, 1557.
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258.
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhofer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018, 556, 244–248.
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 182.
- Wicik, Z.; Eyileten, C.; Jakubik, D.; Simoes, S.N.; Martins, D.C., Jr.; Pavao, R.; Siller-Matula, J.M.; Postula, M. ACE2 Interaction Networks in COVID-19: A Physiological Framework for Prediction of Outcome in Patients with Cardiovascular Risk Factors. J. Clin. Med. 2020, 9, 3743.
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990.
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45.
- Xu, Y.D.; Hua, J.; Mui, A.; O’Connor, R.; Grotendorst, G.; Khalil, N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, 527–539.
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428.
- Wang, L.; Wang, Z.; Liu, J.P. Identification of peptidomimetic telomere dysfunction inhibitor (TELODIN) through telomere dysfunction-induced foci (TIF) assay. STAR Protoc. 2021, 2, 100620.
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83.
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 2016, 7, 11190.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189.

