Differing from such pulmonary stem cells at the distal end of the bronchus as the clublike stem cells
[11], distal airway stem cells (DASC)
[11][12][11,12] and bronchioalveolar stem cells (BASC)
[13], AEC2 stem cells reside within the alveolar epithelium with a significant heterogeneity with variable levels of the marker CD44, telomerase activity, and proliferative and differentiation potentials in response to a variety of cellular signalling in ageing
[14][15][14,15]. Whereas chemically defined EGF/NOGGIN medium supports the basal proliferation of clonal AEC2 cells in the organoids
[16], potentially through a constitutive activity of the single Wnt5a-signalling stromal fibroblast niche, an injury-associated activation of AEC2 stem cell expansion involves autocrine Wnt signalling associated with a potential checkpoint regulation in lieu of the niche juxtacrine Wnt signalling
[17]. Responding to a transient interstitial macrophage-derived IL-1beta, the AEC2 stem cell population undergoes a reshuffle of a distinct subpopulation (damage-associated transient progenitors, DATPs) expressing Il1r1 for alveolar regeneration via a HIF1alpha-mediated glycolysis pathway
[18]. However, in contrast to a full differentiation to AEC1 cells
[16][18][19][16,18,19], AEC2 cell differentiation to AEC1 cells is hampered at an intermediate, partially differentiated status of AEC2 stem cells (called pre-alveolar type-1 transitional cell state, PATS or alveolar differentiation intermediate, ADI) under persistent inflammatory stress conditions, showing cellular replicative senescence in pulmonary fibrosis of both animal models and patients
[4][18][19][20][21][22][23][4,18,19,20,21,22,23]. These findings of AEC2 stem cell senescence-associated differentiation disorder (SADD) may have significant implications in not only the impaired AEC1 cell replenishment during alveolar epithelial repair, but also aberrant trans-differentiation during the pathogenic development of pulmonary fibrosis (
Figure 1).
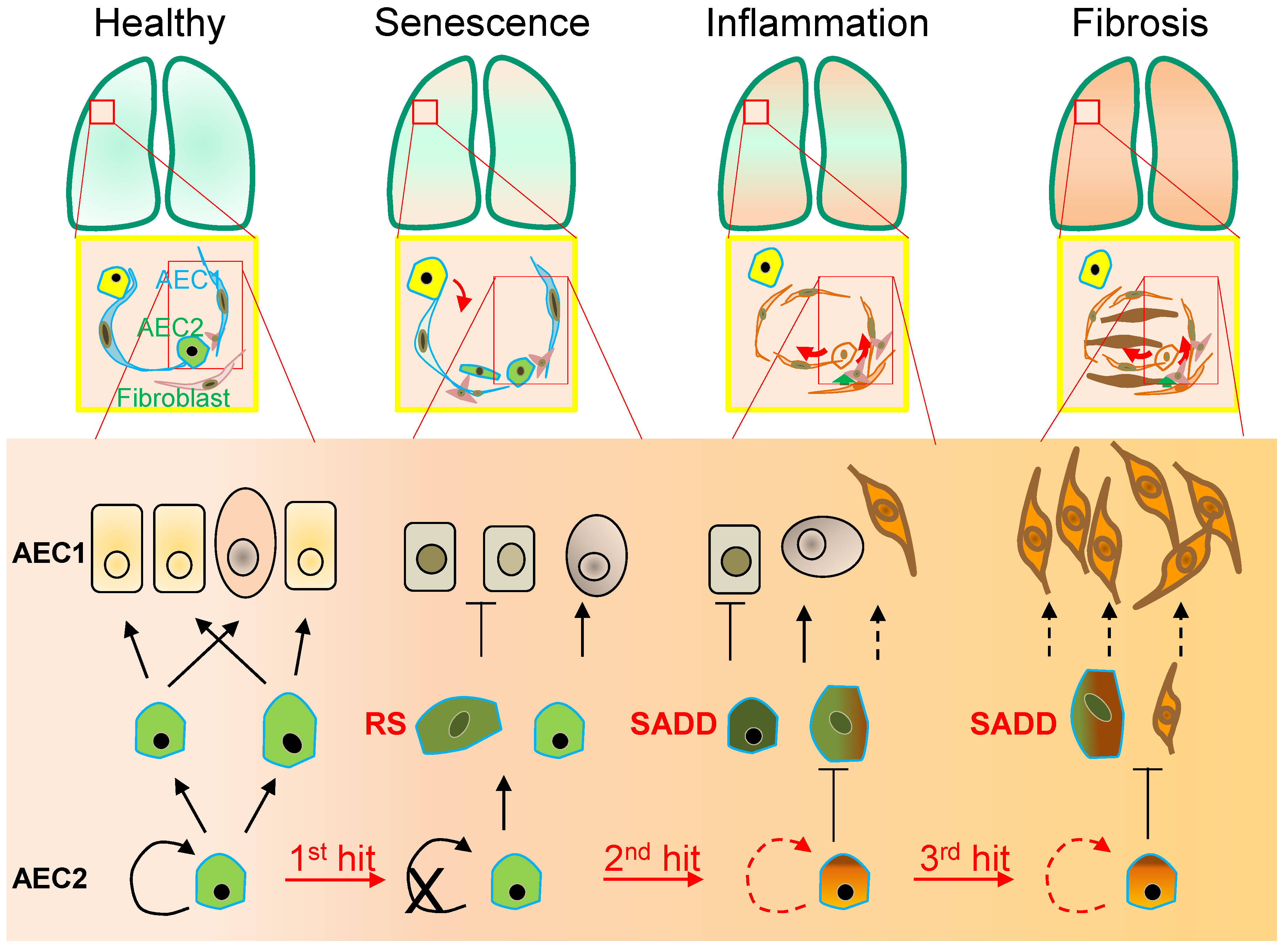
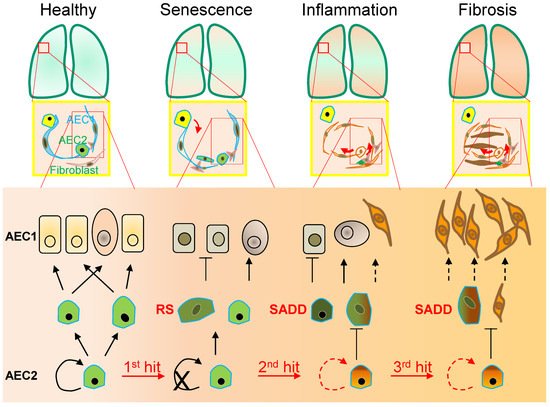
Figure 1. Alveolar monolayer squamous epithelial type 2 (AEC2) stem cell differentiation arrest and transdifferentiation disorder in pulmonary fibrosis. During pulmonary fibrogenesis, AEC2 stem cells are susceptible to stress assaults triggering telomeric DNA damage response (DDR) and replicative senescence and senescence-associated cease of the directional differentiation to alveolar monolayer squamous epithelial type 1 (AEC1) cells. Chronic stress induces senescent AEC2 stem cells to undergo transdifferentiation. The senescence-associated differentiation disorders (SADDs) contribute to myofibroblast proliferation under the condition of senescence-associated low grade inflammation (SALI).
Cellular replicative senescence occurs in various forms depending on the mechanisms leading to replicative senescence. Such forms have been described as stress-induced premature senescence (SIPS), oncogene-induced replicative senescence (OIS), developmental programmed senescence (DPS) and therapy-induced senescence (TIS), which display a general underlying mechanism of the permanent cell cycle arrest in cellular mitotic divisions. Initially referred to as the irreversible cell proliferation ability after a limited number of continuous population doublings for human diploid fibroblasts (HDFs), cell senescence occurs as a general feature of the different types of cell replicative senescence to the resistance to both cell proliferation and cell death signalling, e.g., without responding of mitosis to growth factor induction
[24][25][24,25]. Thus, the primary cellular mechanism of cell replicative senescence converges on the cell cycle permanent arrest provoked by significant stress insults along with sustained DNA damage in response to a variety of physical, chemical and biological stimuli (
Figure 1). Although a variety of diverse stressors accelerate premature terminations of the cell cycle, including chemical toxins, antibiotics and oxidative stress in the airways
[24][26][27][28][29][24,26,27,28,29], cell replicative senescence appears irreversible as demonstrated in SIPS and OIS
[30]. With the enhanced mitogenic signalling such as activation of the Ras/MAPK pathway, and the epigenetic modifications such as histone-3 lysine-9 trimethylation (H3K9Me3) in the senescence-associated heterochromatin foci
[30][31][30,31], OIS occurs to serve as an oncogenic checkpoint to prevent tumor cell clonal expansion and micro-evolutionary transformation with increased oncogenic proteins (such as c-myc
[32] and mTOR
[33]). In contrast to OIS, SIPS and VIS occur with excessive damages to serve as a proliferative and differentiative checkpoint to prevent cellular damage from passing onto daughter cells
[3][34][35][3,34,35].
Intriguingly, the intrinsic reprograming mechanism of a potential cell transition from ceasing replication to entering senescence may involve the transcriptional repression of the
telomerase reverse transcriptase (
TERT) gene expression, conferring cell replicative senescence on oncogene-stimulated cell division
[36][42]. Additionally, TP53 phosphorylation at S15 residue
[37][38] and TP53 fluctuation in oscillatory dynamics
[38][43] may also be involved in the pathway switching from cell replicative senescence to a cell proliferative state. Furthermore, DNA methylation of the Oct4A enhancers may regulate the TP53-dependent TIS through tuning alternative splicing
[39][44]. With an instrumental non-cell-autonomous role of SASP
[40][41][42][41,45,46], cell replicative senescence plasticity represents a hotspot of further investigation in terms of autocrine/juxtacrine/paracrine actions of growth factors, proteases, cytokines, chemokines and extracellular matrix (ECM) components
[43][44][45][46][47][47,48,49,50,51]. In this regard, studies showed that the extracellular vesicles (EVs)—exosomes, ectosomes, microvessels and microparticles—are increased in cell replicative senescence responding to ionizing radiation, chemical reagents or overexpression of oncogenes in a number of cell types including epithelial cells
[48][49][52,53], and that TP53 activation increases EV release and mediates prostate cancer cell senescence induced by telomere shortening or radiation
[49][53], suggesting that extracellular vesicular signalling represents a potential mechanism shaping cellular responses to and from replicative senescence.
2. Senescence-Associated Differentiation Disorder (SADD)
As the lung peripheral tissue stem cells that are essentially required for alveolar epithelial damage repair and regeneration
[10][11][12][13][10,11,12,13], AEC2 stem cells are compromised in replenishing AEC1 cells in pulmonary fibrosis
[23][50][51][23,54,55]. In a long-term process of AEC2 cell self-renewal and directional differentiation to form AEC1 cells by pedigree tracing
[10], AEC2 cells respond to chronic stress with the phenotypes of permanent termination of the cell cycle and differentiation to AEC1
[9][18][19][22][9,18,19,22] by an unclear molecular mechanism
[18][19][20][21][22][23][18,19,20,21,22,23]. In the case of diffuse alveolar damage, in addition to discontinued differentiation to AEC1 cells, AEC2 stem cells arrested at G
2/M of the cell cycle can express a high level of KRT8, and exhibit the cubic and partial spreading morphologies
[18][19][20][21][23][52][53][18,19,20,21,23,56,57]. Moreover, the AEC2 senescent cells appeared to be undergoing a program of transdifferentiation to a mesenchymal state in association with increased levels of α-SMA and collagen
[35][54][55][56][57][58][59][35,58,59,60,61,62,63].
In addition to the discontinued AEC2-to-AEC1 cell differentiation in SADD of IPF, AEC2 cell transdifferentiation in the pathological fibrogenic condition
[35][54][55][56][57][58][59][35,58,59,60,61,62,63] may involve epithelial-mesenchymal transition (EMT)
[58][59][60][61][62][63][64][65][62,63,71,72,73,74,75,76]. Evidence of AEC2 cell contributing to fibrogenesis via transdifferentiation includes co-localization of SPC with α-SMA, Col1α1 and hydroxyproline in the mouse models of pulmonary fibrosis
[35][57][58][59][35,61,62,63]. In addition, recent single-cell RNA sequencing studies revealed that a cell population of replicative senescence is associated with transdifferentiation in the profiles of KRT5
−/KRT17
+ marks and increased gene expressions of a subset of mesenchymal genes such as Col1α1 in the fibrotic lung of patients
[52][53][56,57]. Lineage-tracing studies in mice show that AEC2 stem cells undergo aberrant cellular remodeling with stretching morphological alterations in association with replicative senescence characteristic of SADD, by a mechanism involving increased TGF-
β and TP53 signaling
[19][37][66][19,38,77]. Thus, it is conceivable that SADD underlies the discontinuation of AEC2 stem cell differentiation to AEC1 cells and initiations of myofibroblast activation and matrix deposition under the conditions of alveolar senescence-associated low-grade inflammation (SALI)
[67][68][69][70][78,79,80,81] with inflammatory cell and cytokine signaling
[19][58][59][60][61][62][63][64][65][71][72][19,62,63,71,72,73,74,75,76,82,83]. Further studies are required to investigate if cell replicative senescence may dictate SADD through intracellular mechanisms prior to SALI (
Figure 2), so inhibiting cell replicative senescence may impede SADD as to prevent fibrogenesis in the animal models of telomere dysfunction-induced pulmonary fibrosis
[35].
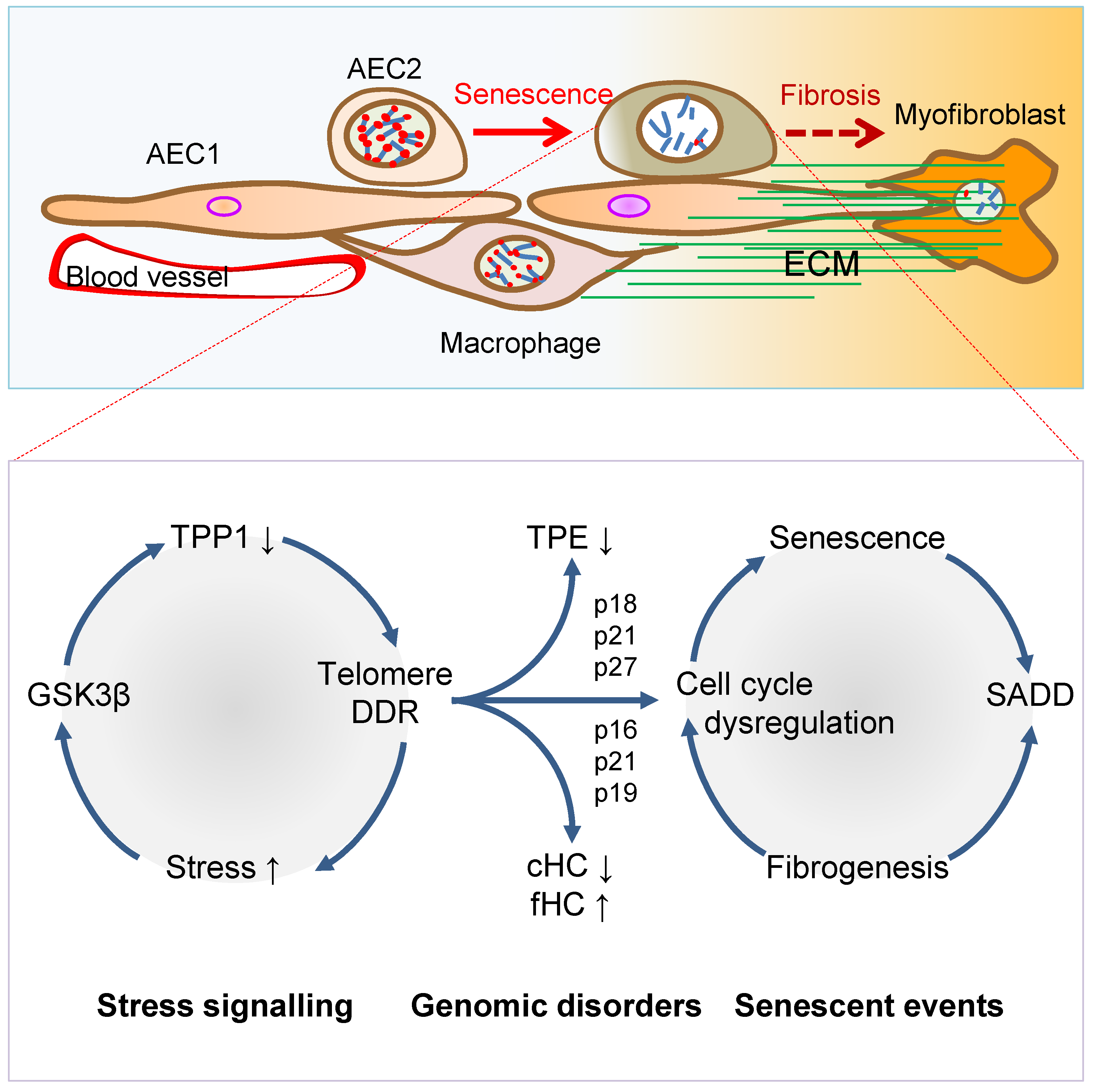
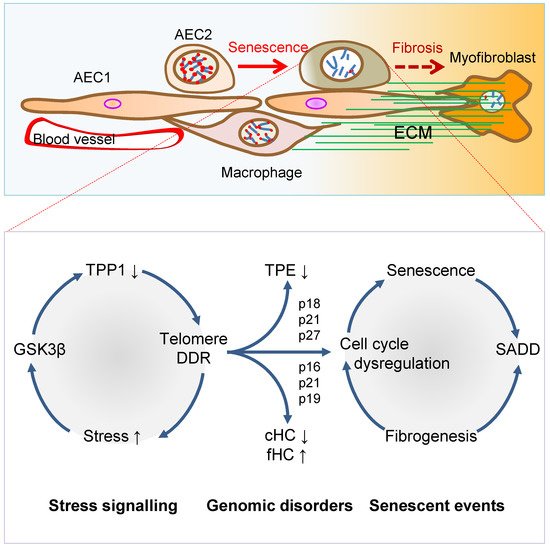
Figure 2. Mechanisms of AEC2 stem cell senescence and SADD. Cellular stress signalling triggers GSK3β-targeting of telomere shelterin complex, inducing the telomerase recruitment protein TPP1 phosphorylation, subjecting phosphorylated TPP1 multisite polyubiquitination and degradation, resulting in telomere uncapping. The telomere uncapping triggers telomeric DDR, resulting in activation of the cyclin-dependent protein kinase inhibitors and cell cycle deregulation through telomere position effect (TPE) and altered constitutive and facultative heterochromatins (cHC and fHC). Unresolved telomeric DNA repair and cell cycle arrest result in stem cell senescence and subsequent SADD including differentiation arrest and trans-differentiation underlying pulmonary fibrosis.
3. Telomere Dysfunction Mediates Pulmonary Senescence and Fibrosis
AEC2 stem cell replicative senescence has been shown to involve DDR in SADD and IPF
[35][73][74][75][76][35,84,85,86,87] under SALI, which is subserved by senescence-associated secretory phenotype (SASP) and infiltrations of inflammatory and immune cells
[67][68][69][70][78,79,80,81] (
Figure 1 and
Figure 2). Genetically, loss of function mutations of the genes encoding
TERT,
telomerase RNA component (
TERC),
poly-(A)-specific ribonuclease (
PARN) and
regulator of telomere length 1 (
RTEL1) underpins patient familial recessive hereditary IPF
[2][18][77][78][79][80][81][82][2,18,88,89,90,91,92,93]. Notably, over a third of IPF cases with hereditary susceptibility of autosomal recessive gene damages could be related to telomere-related gene mutations including telomerase catalytic subunit
TERT and RNA subunit
TERC genes
[6][35][82][83][84][85][86][6,35,93,94,95,96,97], and some could be related to mutations of the genes coding SPC or SPA of alveolar stem cells
[87][88][89][98,99,100]. With the hypothesis that genetic susceptibility involves vulnerable genetic elements such as telomeres predisposing increased pulmonary sensitivity to environmental hazard-accelerated damages
[35], it has been shown that stress-induced DDR occurs rapidly to telomeres, in a more abundant scale in telomeres as compared with non-telomeric regions in the genome
[35][90][91][35,101,102]. The telomeric DDR takes place irrespective of telomerase activity, resistant to DNA repair, rendering irreparable DDR to be persistent in causing cellular replicative senescence
[92][93][94][95][103,104,105,106]. Thus, preventing stress-induced telomere damages may protect the cells from entering replicative senescence and fibrogenesis.
Environmental factors triggering pulmonary senescence and fibrotic disease onset have been a hot topic in aiming to prevent the incurable disease IPF without yet effective therapy
[80][85][91,96]. Since the 1980s, environmental stress factors that drive pulmonary fibrotic onset include smoking, bacterial toxin bleomycin
[96][97][107,108], ionizing radiation (IR)
[98][99][100][109,110,111], and oxidative metabolite reactive oxygen species
[73][101][102][103][104][84,112,113,114,115].
In the telomere syndrome traits, IPF appeared more common than bone marrow diseases
[54][84][105][58,95,127] (such as aplastic anemia
[106][128] or dyskeratosis congenita (DC)
[107][108][109][110][129,130,131,132]). The incidence of IPF is related to the aggravation of lung epithelial injuries, alveolar stem cell replicative senescence and SADD induced by environmental stress factors such as smoking, infection, radiation injury, oxidative stress
[16][35][82][111][16,35,93,117].
3.1. Telomerase Gene Deficiency in Ageing-Related Disorders
Telomerase replenishes telomeres to counteract shortening by
TERC template reverse transcription
[112][113][169,170]. Telomerase catalytic subunit TERT determines telomerase activity in lengthening telomeres, conferring the ability of continuous proliferation and survival on stem cells and tissue precursor cells
[114][115][116][117][118][119][171,172,173,174,175,176]. In addition to elongating telomeres, TERT is involved in maintaining telomere heterochromatin and synthesizing double-stranded RNA through RNA-dependent RNA polymerase activity (RdRP)
[120][121][122][177,178,179]. Telomerase deficiency due to
TERT mutations results in stem cell replicative senescence and exhaustion, tissue fibrosis, aplastic anemia, and skin diseases of premature ageing
[35][67][110][123][124][35,78,132,180,181].
3.2. TGF-β Signaling to Telomerase TERT Gene Repression
TGF-β signaling is closely involved in pulmonary fibrosis
[19][20][22][23][52][53][71][125][126][127][19,20,22,23,56,57,82,199,200,201]. The mechanisms of how TGF-
β family cytokines regulate the intracellular events mediating pulmonary fibrogenesis remain unclear
[128][202]. Studies indicate that mediating the interactions between AEC2 stem cells and fibroblasts, TGF-
β released from cells
[129][203] induces Sonic Hedgehog (SHH) pathway in alveolar epithelial cells by autocrine mechanisms, whereas SHH induces TGF-
β in lung fibroblasts stimulating myofibrosis by paracrine mechanisms, suggesting that re-emergence of SHH in epithelial cells mediates TGF-
β signaling and induces myofibroblast differentiation in a Smoothened receptor-dependent manner with subsequent transcription factor Gli1 activation of the
α-SMA promoter
[130][204].
3.3. Stress-Induced TPP1 Degradation and Telomere Uncapping
In the regulation of telomerase lengthening of telomeres, recruiting telomerase to telomeres represents a most effective step in telomere maintenance.
TheOur recent studies show an intertwined relationship of TPP1 capping of telomeres, recruitment of telomerase, deficiency-induced telomere uncapping, pulmonary senescence, and fibrosis
[35][131][35,213].
4. Targeted Intervention of Cellular Senescence and Tissue Fibrosis
Since cellular replicative senescence plays a causal role in tissue remodelling, it appears particularly appealing to determine if elimination or prevention of cellular replicative senescence may provide beneficial outcomes to mitigate pathogenic tissue remodelling and ageing-related disorders under various disease conditions. Considerable evidence suggests that targeted removal or prevention of cell replicative senescence alleviates SADD in fibrosis, including VIS or stress-induced fibrosis
[3][35][3,35].
4.1. Targeting Anti-Apoptotic Gene Bcl-2 to Clear Senescent Cells
Since replicative senescence cells have upregulated anti-apoptotic proteins Bcl-w and Bcl XL which underpin senescent cells’ antiapoptotic ability with long-term survival, drugs that inhibit Bcl survival proteins have been studied and named as senolytics for removing replicative senescence cells
[132][133][217,218]. Although the U.S. Food and Drug Administration (FDA) approved the selective Bcl-2 inhibitor venetoclax (abt-199) used in leukemia, it has not had a significant effect on anti-ageing tests in vitro. Compound screening-identified that its homologue navitoclax (abt-263) effectively inhibits the effects of Bcl-2, Bcl XL and Bcl-w, and induces apoptosis and thus clearance of replicative senescence cells
[132][217].
4.2. Targeting TP53 and p16
INK4A
Tumor Suppressor Genes to Clear Senescent Cells
Recent studies have consistently shown that senescent cells can be eliminated by targeting the cells with high levels of p16
INK4A and TP53 tumor suppressor gene expressions. Because p16
INK4a is significantly increased in senescent cells, AP20187 that targets FK506 using a minimal p16
INK4a promoter element triggers a dimer formation of FK506 binding protein and caspase-8 to effectively induce senescent cell apoptosis, resulting in a significant reduction of the incidence and mortality of cardiovascular diseases in aged mice
[134][221].
4.3. Preventing Telomere Dysfunction and Pulmonary Fibrosis by TELODIN
By screening a peptide library, an 8-mer peptide (TELODIN) that significantly inhibited telomere dysfunction was discovered
[131][213]. Corresponding to the
β-turn hairpin-like blade 7 of FBW7 E3 ubiquitin ligase WD40 domain, as synthesized in a native or retro-inverso (reversed, inversed in dextral amino acids) configuration, TELODIN competitively inhibited stress-induced TPP1 accelerated turnover, telomere shortening and pulmonary fibrosis once applied through intratracheal instillation in mice
[35][131][35,213].
5. Conclusions
Pulmonary ageing and fibrosis occur in association with AEC2 stem cell senescence and SADD. AEC2 stem cell failure of differentiation to AEC1 cells in epithelial damage repair, and trans-differentiation to mesenchymal fibrogenic remodeling, are the two critical cellular processes mediated by telomere dysfunction. Downstream of stress-induced uncapping of telomeres or TGF-
β-signaling repression of the telomerase
TERT gene, AEC2 stem cell senescence drives alveolar epithelial fibrogenesis, representing a key molecular target for intervention. Preventing AEC2 stem cell senescence by promoting telomere capping integrity using TELODIN, or removing senescent cells by promoting apoptosis using senolytics, is emerging as a promising strategy in the studies of pulmonary fibrosis intervention.