 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kazuhiko Uchida | + 2362 word(s) | 2362 | 2022-03-14 05:26:11 | | | |
| 2 | Rita Xu | Meta information modification | 2362 | 2022-03-15 05:10:45 | | | | |
| 3 | Rita Xu | + 88 word(s) | 2450 | 2022-03-15 05:12:01 | | |
Video Upload Options
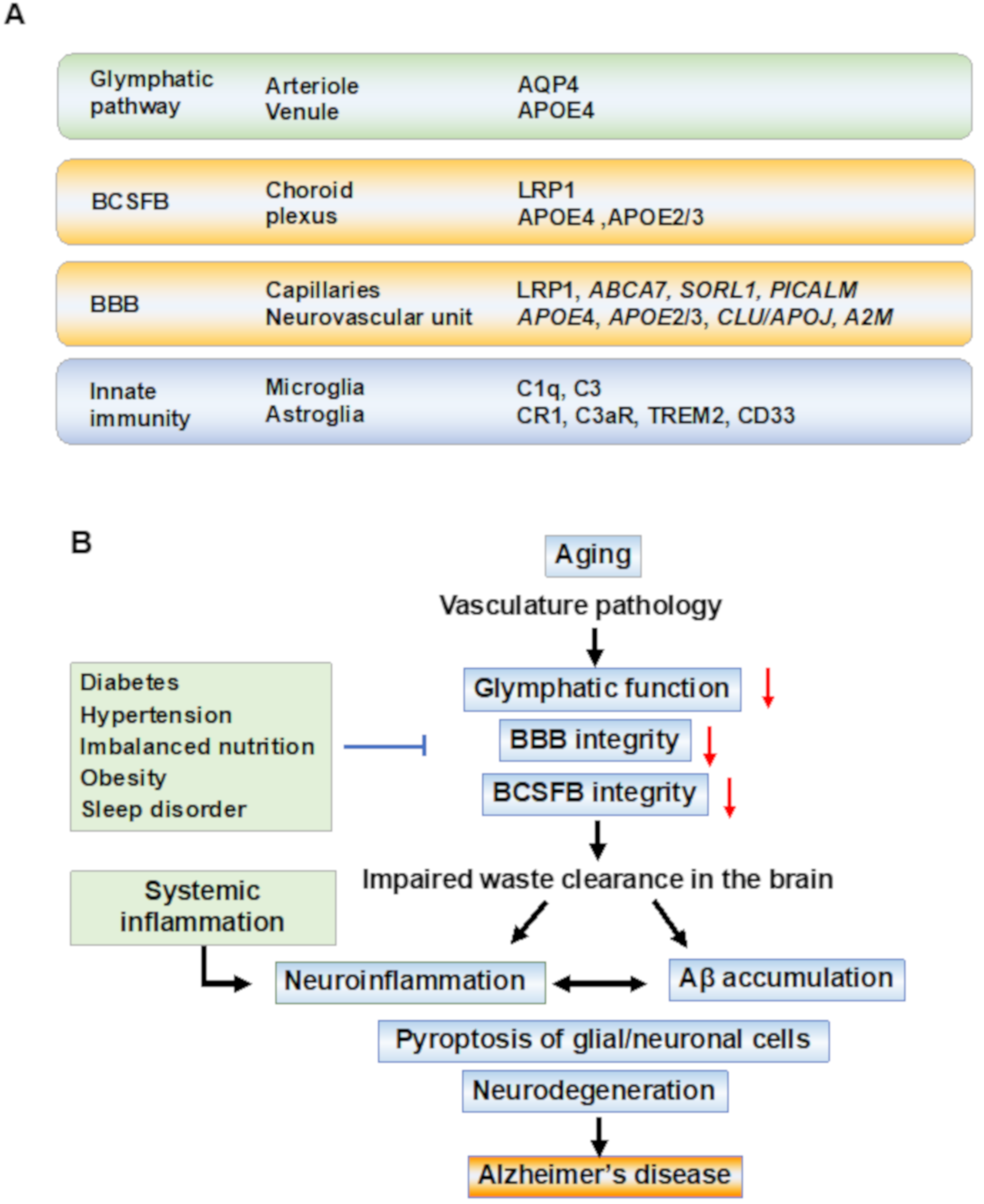
Alzheimer’s disease (AD) is a multifactorial disease with a heterogeneous etiology. The pathology of Alzheimer’s disease is characterized by amyloid-beta and hyperphosphorylated tau, which are necessary for disease progression. Many clinical trials on disease-modifying drugs for AD have failed to indicate their clinical benefits. Recent advances in fundamental research have indicated that neuroinflammation plays an important pathological role in AD. Damage- and pathogen-associated molecular patterns in the brain induce neuroinflammation and inflammasome activation, causing caspase-1-dependent glial and neuronal cell death. These waste products in the brain are eliminated by the glymphatic system via perivascular spaces, the blood-brain barrier, and the blood–cerebrospinal fluid barrier. Age-related vascular dysfunction is associated with an impairment of clearance and barrier functions, leading to neuroinflammation. The proteins involved in waste clearance in the brain and peripheral circulation may be potential biomarkers and drug targets in the early stages of cognitive impairment.
1. Introduction
2. Multifactorial Pathobiology in AD
2.1. The Danger Signal Activates Neuroinflammation and Induces Pyroptosis
2.2. The Glymphatic System for Clearance of Brain Waste
2.3. BBB and Blood–CSF Barrier in AD Pathobiology
2.4. Microglial Activation for Waste Clearance in the Brain
References
- GBD 2016 Dementia Collaborators. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106.
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021.
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular Dysfunction-The Disregarded Partner of Alzheimer’s Disease. Alzheimers Dement. 2019, 15, 158–167.
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2021. Alzheimers Dement. 2021, 7, e12179.
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771.
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17.
- Arai, T.; Hasegawa, M.; Nonoka, T.; Kametani, F.; Yamashita, M.; Hosokawa, M.; Niizato, K.; Tsuchiya, K.; Kobayashi, Z.; Ikeda, K.; et al. Phosphorylated and Cleaved TDP-43 in ALS, FTLD and Other Neurodegenerative Disorders and in Cellular Models of TDP-43 Proteinopathy. Neuropathology 2010, 30, 170–181.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128.
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 280–292.
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Beta-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774.
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446.
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109.
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in Neuroinflammatory and Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10248.
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β Induces NLRP1-Dependent Neuronal Pyroptosis in Models of Alzheimer’s Disease. Cell Death Dis. 2014, 5, e1382.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2012, 493, 674–678.
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear Factor-Kappa β as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2019, 150, 113–137.
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572.
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88.
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do Infections Have a Role in the Pathogenesis of Alzheimer Disease? Nat. Rev. Neurol. 2020, 16, 193–197.
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes Simplex Virus Type 1 DNA Is Located within Alzheimer’s Disease Amyloid Plaques. J. Pathol. 2009, 217, 131–138.
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-Negative Bacterial Molecules Associate with Alzheimer Disease Pathology. Neurology 2016, 87, 2324–2332.
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.-G.; Elgh, F. Reactivated Herpes Simplex Infection Increases the Risk of Alzheimer’s Disease. Alzheimers Dement. 2015, 11, 593–599.
- Butler, L.; Walker, K.A. The Role of Chronic Infection in Alzheimer’s Disease: Instigators, Co-Conspirators, or Bystanders? Curr. Clin. Microbiol. Rep. 2021, 8, 199–212.
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s Disease: Possible Role of Periodontal Diseases. Alzheimers Dement. 2008, 4, 242–250.
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; de Leon, M.J. Periodontal Disease as a Possible Cause for Alzheimer’s Disease. Periodontol. 2000 2020, 83, 242–271.
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas Gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333.
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and Central Immune System Crosstalk in Alzheimer Disease—A Research Prospectus. Nat. Rev. Neurol. 2021, 17, 689–701.
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462.
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic Inflammation Induces Apoptosis with Variable Vulnerability of Different Brain Regions. J. Chem. Neuroanat. 2005, 30, 144–157.
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and Systemic Endotoxin Challenges Exacerbate the Local Inflammatory Response and Increase Neuronal Death during Chronic Neurodegeneration. J. Neurosci. 2005, 25, 9275–9284.
- Nedergaard, M.; Goldman, S.A. Glymphatic Failure as a Final Common Pathway to Dementia. Science 2020, 370, 50–56.
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of Amyloid Beta-Protein Clearance and Alzheimer’s Disease Susceptibility by the LDL Receptor-Related Protein Pathway. J. Clin. Investig. 2000, 106, 1159–1166.
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological Amyloid-Beta Clearance in the Periphery and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2015, 130, 487–499.
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired Glymphatic Function and Clearance of Tau in an Alzheimer’s Disease Model. Brain 2020, 143, 2576–2593.
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111.
- Nedergaard, M. Garbage Truck of the Brain. Science 2013, 340, 1529–1530.
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann. Neurol. 2014, 76, 845–861.
- Zhou, Y.; Cai, J.; Zhang, W.; Gong, X.; Yan, S.; Zhang, K.; Luo, Z.; Sun, J.; Jiang, Q.; Lou, M. Impairment of the Glymphatic Pathway and Putative Meningeal Lymphatic Vessels in the Aging Human. Ann. Neurol. 2020, 87, 357–369.
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024.
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302.
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood-Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76.
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate Immunity in Alzheimer’s Disease. Nat. Immunol. 2015, 16, 229–236.
- Shi, Y.; Holtzman, D.M. Interplay between Innate Immunity and Alzheimer Disease: APOE and TREM2 in the Spotlight. Nat. Rev. Immunol. 2018, 18, 759–772.
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Investig. 2017, 127, 3240–3249.
- Perry, V.H.; Newman, T.A.; Cunningham, C. The Impact of Systemic Infection on the Progression of Neurodegenerative Disease. Nat. Rev. Neurosci. 2003, 4, 103–112.
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic Inflammation and Disease Progression in Alzheimer Disease. Neurology 2009, 73, 768–774.
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M. Systemic Inflammation Impairs Microglial Aβ Clearance through NLRP 3 Inflammasome. EMBO J. 2019, 38, e101064.
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-Wide Association Study Identifies Variants at CLU and PICALM Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1088–1093.
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–435.
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099.
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic Variation in Aquaporin-4 Moderates the Relationship between Sleep and Brain Aβ-Amyloid Burden. Transl. Psychiatry 2018, 8, 47.
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; De Natale, E.R.; Politis, M. Alzheimer’s Disease Neuroimaging Initiative Aquaporin-4 Polymorphisms Predict Amyloid Burden and Clinical Outcome in the Alzheimer’s Disease Spectrum. Neurobiol. Aging 2021, 97, 1–9.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458.
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and Refinement of Loci Associated with Lipid Levels. Nat. Genet. 2013, 45, 1274–1283.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116.


