Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Kazuhiko Uchida.
Alzheimer’s disease (AD) is a multifactorial disease with a heterogeneous etiology. The pathology of Alzheimer’s disease is characterized by amyloid-beta and hyperphosphorylated tau, which are necessary for disease progression. Many clinical trials on disease-modifying drugs for AD have failed to indicate their clinical benefits. Recent advances in fundamental research have indicated that neuroinflammation plays an important pathological role in AD.
- Aβ clearance
- blood-brain barrier
- glymphatic system
- MCI
- dementia
- biomarker
1. Introduction
Dementia is one of the most economically burdensome diseases in families as it is a long-term illness causing disability in daily life. The number of people with dementia worldwide doubled from 20.2 million in 1990 to 43.8 million in 2016 [1]. According to the World Alzheimer Report 2021, the number of people with dementia worldwide is 55 million and is estimated to reach 78 million by 2030 [2]. Alzheimer’s disease (AD) is the most common type of dementia, followed by vascular dementia, dementia with Lewy bodies, and frontotemporal lobe dementia.
Late-onset AD, which occurs in people aged 65 years and above, has continuous progression from the preclinical stage without clinical symptoms to the pre-dementia stage, mild cognitive impairment (MCI), and AD with disability in daily life. Amyloid beta (Aβ) plaques and tau phosphorylation in the intraneuronal neurofibrillary tangles (NFTs) are major and classic characteristics of AD pathology. Aβ aggregates are neurotoxic and induce neurodegeneration; however, a substantial body of evidence has recently indicated that AD has a heterogeneous etiology and multifactorial pathogenesis [3].
Numerous research and clinical trials targeting Aβ have been performed in recent decades to overcome this devastating disease. There are currently 126 agents in 152 ongoing clinical trials for AD therapeutics, and new types of disease-modifying drugs targeting the underlying pathobiology of AD are also being developed [4]. In 2021, aducanumab, an anti-Aβ human monoclonal antibody, was approved by the United States Food and Drug Administration under the accelerated approval pathway for early AD [5]. However, many clinical trials of immunotherapy-targeted Aβ have failed to develop effective and safe drugs, possibly due to the heterogeneity of etiology and pathobiology in AD.
Neurodegenerative diseases are characterized by the deposition of aggregated proteins such as Aβ, tau, α-synuclein, and the TAR DNA-binding protein of 43 kDa (TDP-43) in the brain [6,7][6][7]. In sporadic AD, the accumulation of Aβ starts more than 20 years before disease onset without any symptoms, owing to impaired waste clearance rather than an overproduction of the peptide [8,9,10][8][9][10]. Recent studies have shown that early intervention is required for providing effective therapy and preventing dementia [11].
2. Multifactorial Pathobiology in AD
2.1. The Danger Signal Activates Neuroinflammation and Induces Pyroptosis
Late-onset AD is the most common type of dementia among elderly individuals. As observed in autopsied AD brains, Aβ, which is produced from the amyloid precursor protein (APP) by the sequential cleavage of β- and γ-secretases, forms senile plaques. It is a major pathogenic factor as its production increases in hereditary AD. An experimental mouse model overexpressing APP demonstrated the loss of synaptic function and AD-like pathophysiology. Aβ42 accumulates in the brain parenchyma, forming plaques, whereas Aβ40 primarily accumulates in the cerebral blood vessel walls, causing amyloid angiopathy. The balance between Aβ production and clearance determines the amount of Aβ accumulation in the brain. An excessive accumulation of Aβ in the brain is observed in AD, whilst Aβ in CSF and peripheral blood is decreased due to an impaired clearance from the brain. The clearance of both Aβ42 and Aβ40 was reduced to 30% in AD individuals compared to that in non-demented controls [10]. APP processing occurs in an age-dependent manner possibly due to the inflammatory response. The imbalance of Aβ clearance and production might contribute to Aβ accumulation in late-onset AD.
Recently, caspase-1 dependent programmed cell death associated with inflammation has been proposed as a new type of cell death, named pyroptosis, which is characterized by inflammasome-related cell death with membrane rupture and cytokine release [14][12]. Several lines of evidence indicated that Aβ aggregates activate the inflammasomes and induce pyroptosis, resulting in glial and neuronal cell death [15,16,17][13][14][15]. Aβ binds to and stimulates the pattern recognition receptors, including the nucleotide oligomerization domain-like receptor proteins, resulting in inflammasome formation by the recruitment of the apoptosis-associated speck-like protein which contains a caspase recruitment domain and procaspase-1. Activated caspase-1 cleaves pro-interleukin (IL)-1β and pro-IL-18 into their mature forms, and cleaves gasdermin D to generate a 31-kDa N-terminal fragment which forms plasma membrane pores.
Several factors other than misfolded protein aggregates such as Aβ and tau prime inflammatory responses and induce caspase-1 dependent pyroptosis in the brain. The priming signals include lipopolysaccharide, bacterial pathogens, toxins, and double-stranded DNA. These pathogen-and host-derived signals induce innate immunity and activate nuclear factor-kappa B (NF-κB) signaling, resulting in the production of Aβ and inflammatory cytokines [18,19][16][17]. Thus, the production of Aβ may be a reactive compensatory response to several “danger signals” for nerve cells, such as pathogen-associated danger patterns (PAMPs) and damage-associated danger patterns (DAMPs) in the brain due to aging [20][18].
The role of infection in AD has been debated for 30 years [21][19]. Recent advances in research have reignited interest and discussion regarding the role of infection in AD. Several types of virus and bacteria including herpesvirus and Porphyromonas gingivalis (P. gingivalis) have been detected in the brain and colocalized with Aβ plaque [22,23][20][21]. Herpes simplex virus 1 (HSV-1) is usually latent in many elderly brains but may be reactivated under certain conditions leading to neuroinflammation with an increased risk of AD [24][22]. Other herpes viruses, including HSV-2, human herpesvirus-3 (HHV-3), and HHV-6, have been investigated for their association with AD [25][23]. P. gingivalis is a key pathogen in periodontitis, and several reports suggest a role of P. gingivalis in AD pathogenesis [26,27,28][24][25][26]. Viral and bacterial pathogens can cross the blood-brain barrier (BBB), and microglia recognize these pathogens and become activated, thus releasing proinflammatory signals in the brain parenchyma. Several lines of evidence supporting crosstalk between the peripheral and central immune systems suggest a role of systemic inflammation in AD [29,30,31,32][27][28][29][30]. More studies are required to address the question of whether chronic infection has a role in the disease progression of AD and whether the modulation of the signaling pathway in inflammation improves cognitive function.
2.2. The Glymphatic System for Clearance of Brain Waste
Recent studies on brain waste clearance have revealed that the impaired glymphatic system and dysfunction of the blood–CSF barrier (BCSFB) and BBB resulted in the accumulation of waste following neuroinflammation in AD pathogenesis [13,33,34,35][31][32][33][34]. Nedergaard and colleagues demonstrated that CSF moves into the parenchyma through the glymphatic system [12,36][35][36]. They showed that CSF enters the parenchyma through the periarterial pathway and flows towards the veins. The movement of periarterial CSF into the parenchyma facilitates the clearance of waste, such as Aβ, in interstitial fluid (ISF) into the paravenous space. The interexchange of CSF and ISF is crucial for brain homeostasis and waste clearance in the parenchyma. CP secretes CSF into the cerebral ventricles. Located in the brain ventricles, CP forms a BCSFB consisting of tightly connected epithelial cells, pericytes, and stromal space surrounding the capillaries and connective tissue.
CSF fluxes by the glymphatic paravascular system are reduced by aging. In an analysis using radiolabeled Aβ, the influx and efflux of CSF decreased in aged mice compared to young mice [37]. Cerebral vasculature aging is associated with CSF–ISF flux in the glymphatic system [38]. Arterial stiffness with aging leads to a reduction in arterial pulsatility, which drives the periarterial CSF into the parenchyma. The genetic deletion of aquaporin-4 water channels, expressed at the vascular endfeet of astrocytes, reduces glymphatic function and Aβ clearance by approximately 50% [12][35]. Aging is partly associated with the loss of perivascular aquaporin-4 polarization, resulting in the impairment of CSF–ISF exchange [37]. Failure of the glymphatic system due to aging reduces waste clearance in the parenchyma and results in the accumulation of Aβ aggregates and phosphorylated tau (p-tau), which induce neuroinflammation [13,36,39,40][31][36][39][40].
2.3. BBB and Blood–CSF Barrier in AD Pathobiology
Healthy brain function is dependent on healthy cerebral blood vessels and blood flow which supply oxygen and glucose. Oxygen and glucose transport across the BBB are a neurovascular coupling mechanism. The BBB is formed by a layer of capillary endothelial cells coupled with tight junctions, pericytes, smooth muscle cells, and astrocyte endfeet. To maintain brain homeostasis, BBB separates the parenchyma from the blood and prevents the infiltration of pathogens, blood cells, and neurotoxic components into the brain.
The physiology of molecular transport across the BBB and its dysfunction in neurological disorders have been intensively studied. Recent neuroimaging studies using dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) to quantify the regional BBB permeability revealed BBB breakdown in the hippocampus of individuals with early cognitive dysfunction [41] and aging [42]. BBB breakdown contributes to cognitive decline in apolipoprotein E (APOE)-4 carriers by Aβ-independent pathology [43]. Increased BBB permeability allows the infiltration of blood-derived macromolecules, leukocytes, hemoglobin, and neurotoxic agents produced by the pathogens. Because of BBB breakdown, free radicals are produced and proteases are released from leukocytes into the parenchyma. These pathogen- and host-derived “danger” signals lead to neuroinflammation in AD pathobiology.
2.4. Microglial Activation for Waste Clearance in the Brain
Microglia are present in the brain, forming a major component of the innate immune system. Increasing evidence supports the role of peripheral and central innate immune responses in neurodegenerative diseases [44,45][44][45]. Microglia express pattern recognition receptors, which bind and respond to PAMPs and DAMPs, including Aβ species. Activated microglia reduce Aβ accumulation by phagocytosis in a complement protein-dependent manner. Depending on the circumstances, microglia activation demonstrates a divergent response. Beneficial outcomes by increased clearance of debris with no or limited damage to neurons and detrimental outcomes can be observed by producing pro-inflammatory cytokines, leading to chronic inflammation and neurodegeneration [46]. There is cumulative evidence on an innate immune response suggesting peripheral and central crosstalk in AD pathogenesis [47,48][47][48]. Systemic inflammatory events are associated with cognitive decline in several cohorts and brain-resident macrophages and microglia are activated with the release of cytokines by the peripheral administration of lipopolysaccharides in mice models [30,49][28][49]. Thus, there is crosstalk between the peripheral and central immune systems. The inflammatory signals communicate with the central nervous system across BBB and BCSFB. The breakdown of these barriers activates innate immunity and induces neuroinflammation as normal responses in the preclinical stages of AD.
Genome-wide association studies (GWAS) for late-onset AD have identified several variants associated with the immune system, lipid metabolism, and synaptic function. A series of GWAS studies identified susceptibility genes, and several of these genes are involved in the regulation of waste clearance systems (Figure 1). The glymphatic CSF–ISF flow is driven by CSF influx in the periarterial space of major cerebral arteries and efflux to the perivascular space of venules. CSF–ISF drains into the meningeal lymphatic vessels and cervical lymph nodes. Arterial pulsatility and CSF flow are believed to drive ISF into the perivenous space by the hydrostatic pressure and osmotic gradients. APOE4 is a major genetic risk factor for AD which leads to BBB dysfunction in mice models and humans. After identifying APOE4 as a genetic risk factor for AD, CLU (also known as APOJ), CR1, PICAM, ABCA7, and CD33 were identified [50,51,52][50][51][52]. Astrocytic water channel aquaporin-4 (AQP4) plays a pivotal role in the glymphatic system, and AQP4 variants were associated with Aβ burden and an increased risk of AD [53,54][53][54]. Several association studies on genetic variations of low-density lipoprotein receptor-related protein 1 (LRP1) suggested that LRP1 variants may not influence AD risk [55] but were associated with the lipid levels [56]. Genetic meta-analysis revealed an association of genetic variations with the immunity and lipid metabolism, including CD33 and sortilin-related receptor-1 (SORL1) (also known as the low-density lipoprotein receptor relative with 11 ligand-binding repeats, LR11) [57]. Triggering receptor expressed on myeloid cells 2 (TREM2) carrying a rare variant (p.Arg47His) which is associated with three- to four-fold increased risk of developing AD [58,59][58][59].
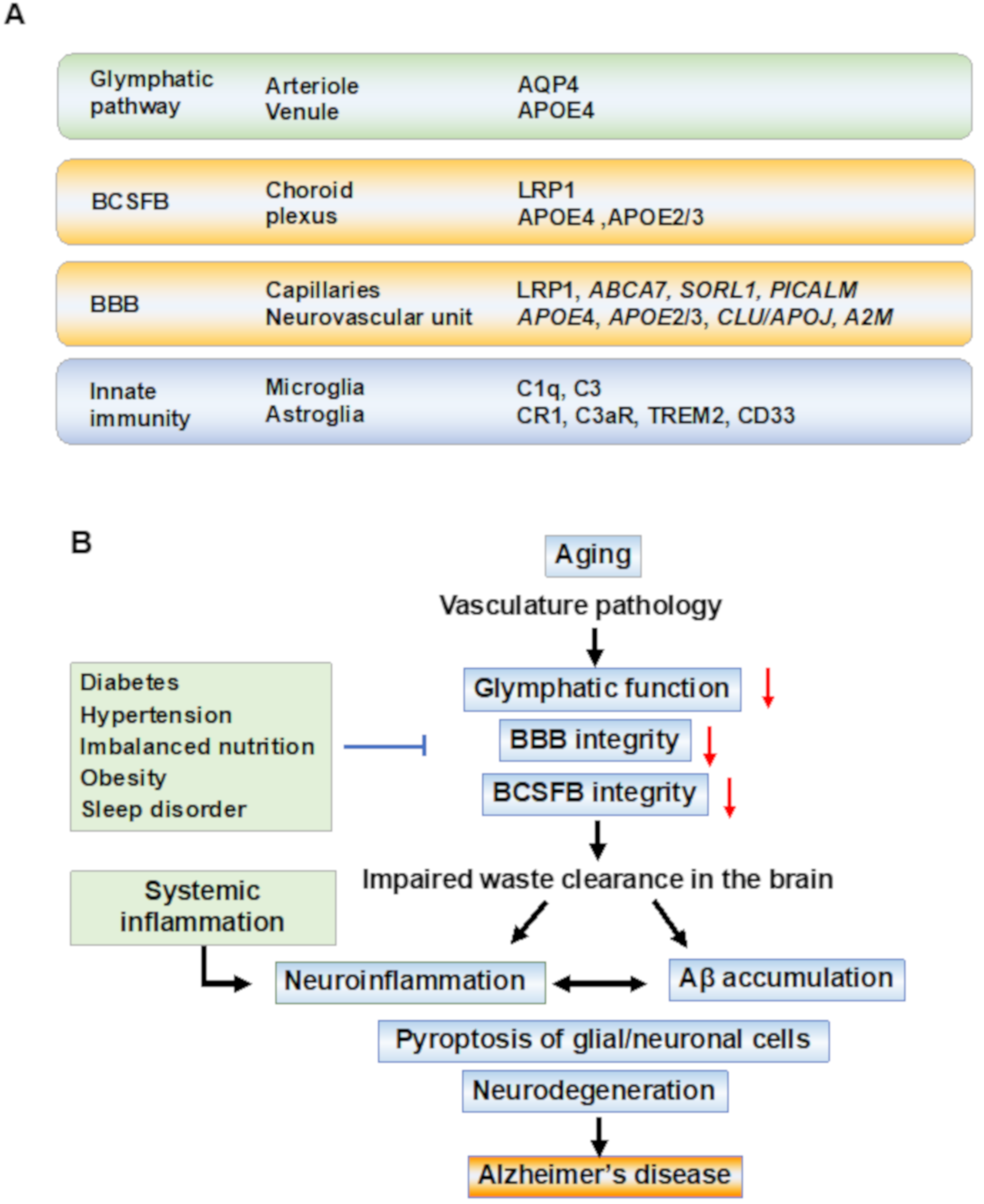
Figure 1. Waste clearance systems and pathobiological events in Alzheimer’s disease. (A) Anatomical regions and key molecules involved in waste clearance systems. Anatomical regions and key molecules identified in the genome-wide association study (GWAS) associated with Alzheimer’s disease (AD) are shown. The molecules which have genetic variations associated with an increased risk for AD are shown as identified by GWAS. The paravascular glymphatic flow is dependent on the water channel aquaporin-4 (AQP4) at the astrocytic endfeet. The blood–cerebrospinal fluid barrier (BCSFB) at the choroid plexus and the blood-brain barrier (BBB) at the capillaries regulate the transport of molecules into and out of the brain. Microglia survey the brain to support homeostasis, and the pathogens and amyloid-beta (Aβ) aggregates are cleared by microglia through receptor-mediated phagocytosis. Astrocytes (astroglia) promote neuroinflammation. After exposure to Aβ, astroglia release NF-kB and complement components, which act through the complement receptors such as complement receptor 1 (CR1) and complement component 3a receptor (C3aR). Aβ is removed by the low-density lipoprotein (LDL) receptor-related protein-1 (LRP-1)-mediated transport. The variants of adenosine triphosphate-binding cassette transporter A7 (ABCA7), sortilin-related receptor-1 (SORL1), phosphatidylinositol-binding clathrin assembly (PICALM), clusterin (CLU/APOJ), and alpha-2 macroglobulin (A2M) affect BBB and clearance functions. Among the rare variants of genes associated with an increased risk of AD, triggering receptors expressed on myeloid cells-2 (TREM2), CD33, and CR1 are expressed on microglia and ascribed to innate immune pathways. (B) Underlined mechanisms and pathobiological events in the disease progression of AD. In sporadic AD, the aging of the vasculature may be an initial event in AD pathobiology. The waste clearance pathways contribute to brain homeostasis and its dysfunction is associated with vasculature disease. In preclinical stages, age-related vascular changes including vasomotor dysfunction, structural remodeling, and chronic inflammation, cause the breakage of tight junctions between epithelial cells in the BBB, and barrier dysfunction in the brain leads to the neuroinflammation of the parenchyma. Inflammation-related signal transduction induces beta-site amyloid precursor protein (APP) cleaving enzyme-1 (BACE1) expression, leading to Aβ production following tau pathology, resulting in amyloid plaques and neurofibrillary tangles. The accumulation of Aβ reduces capillary blood flow and impairs waste clearance. Systemic inflammation is a risk factor for AD, and recent studies have suggested that systemic inflammation can drive neuroinflammation. In these aging processes, an increase in damage-associated danger patterns, including Aβ, which has synaptotoxicity in the brain, promotes neuroinflammation in the parenchyma, and finally, caspase-1 dependent programmed cell death (pyroptosis) in the glial and neuronal cells occurs, resulting in cognitive impairment. Abbreviations: AD—Alzheimer’s disease; BBB—blood-brain barrier; BCSFB—blood–CSF barrier.
References
- GBD 2016 Dementia Collaborators. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106.
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021.
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular Dysfunction-The Disregarded Partner of Alzheimer’s Disease. Alzheimers Dement. 2019, 15, 158–167.
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2021. Alzheimers Dement. 2021, 7, e12179.
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771.
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17.
- Arai, T.; Hasegawa, M.; Nonoka, T.; Kametani, F.; Yamashita, M.; Hosokawa, M.; Niizato, K.; Tsuchiya, K.; Kobayashi, Z.; Ikeda, K.; et al. Phosphorylated and Cleaved TDP-43 in ALS, FTLD and Other Neurodegenerative Disorders and in Cellular Models of TDP-43 Proteinopathy. Neuropathology 2010, 30, 170–181.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128.
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 280–292.
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Beta-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774.
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446.
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109.
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in Neuroinflammatory and Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10248.
- Tan, M.-S.; Tan, L.; Jiang, T.; Zhu, X.-C.; Wang, H.-F.; Jia, C.-D.; Yu, J.-T. Amyloid-β Induces NLRP1-Dependent Neuronal Pyroptosis in Models of Alzheimer’s Disease. Cell Death Dis. 2014, 5, e1382.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2012, 493, 674–678.
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear Factor-Kappa β as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2019, 150, 113–137.
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572.
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88.
- Itzhaki, R.F.; Golde, T.E.; Heneka, M.T.; Readhead, B. Do Infections Have a Role in the Pathogenesis of Alzheimer Disease? Nat. Rev. Neurol. 2020, 16, 193–197.
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes Simplex Virus Type 1 DNA Is Located within Alzheimer’s Disease Amyloid Plaques. J. Pathol. 2009, 217, 131–138.
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-Negative Bacterial Molecules Associate with Alzheimer Disease Pathology. Neurology 2016, 87, 2324–2332.
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.-G.; Elgh, F. Reactivated Herpes Simplex Infection Increases the Risk of Alzheimer’s Disease. Alzheimers Dement. 2015, 11, 593–599.
- Butler, L.; Walker, K.A. The Role of Chronic Infection in Alzheimer’s Disease: Instigators, Co-Conspirators, or Bystanders? Curr. Clin. Microbiol. Rep. 2021, 8, 199–212.
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s Disease: Possible Role of Periodontal Diseases. Alzheimers Dement. 2008, 4, 242–250.
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; de Leon, M.J. Periodontal Disease as a Possible Cause for Alzheimer’s Disease. Periodontol. 2000 2020, 83, 242–271.
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas Gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333.
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and Central Immune System Crosstalk in Alzheimer Disease—A Research Prospectus. Nat. Rev. Neurol. 2021, 17, 689–701.
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 2007, 55, 453–462.
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic Inflammation Induces Apoptosis with Variable Vulnerability of Different Brain Regions. J. Chem. Neuroanat. 2005, 30, 144–157.
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and Systemic Endotoxin Challenges Exacerbate the Local Inflammatory Response and Increase Neuronal Death during Chronic Neurodegeneration. J. Neurosci. 2005, 25, 9275–9284.
- Nedergaard, M.; Goldman, S.A. Glymphatic Failure as a Final Common Pathway to Dementia. Science 2020, 370, 50–56.
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of Amyloid Beta-Protein Clearance and Alzheimer’s Disease Susceptibility by the LDL Receptor-Related Protein Pathway. J. Clin. Investig. 2000, 106, 1159–1166.
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological Amyloid-Beta Clearance in the Periphery and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2015, 130, 487–499.
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired Glymphatic Function and Clearance of Tau in an Alzheimer’s Disease Model. Brain 2020, 143, 2576–2593.
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111.
- Nedergaard, M. Garbage Truck of the Brain. Science 2013, 340, 1529–1530.
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann. Neurol. 2014, 76, 845–861.
- Zhou, Y.; Cai, J.; Zhang, W.; Gong, X.; Yan, S.; Zhang, K.; Luo, Z.; Sun, J.; Jiang, Q.; Lou, M. Impairment of the Glymphatic Pathway and Putative Meningeal Lymphatic Vessels in the Aging Human. Ann. Neurol. 2020, 87, 357–369.
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024.
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat. Med. 2019, 25, 270–276.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302.
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood-Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76.
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate Immunity in Alzheimer’s Disease. Nat. Immunol. 2015, 16, 229–236.
- Shi, Y.; Holtzman, D.M. Interplay between Innate Immunity and Alzheimer Disease: APOE and TREM2 in the Spotlight. Nat. Rev. Immunol. 2018, 18, 759–772.
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Investig. 2017, 127, 3240–3249.
- Perry, V.H.; Newman, T.A.; Cunningham, C. The Impact of Systemic Infection on the Progression of Neurodegenerative Disease. Nat. Rev. Neurosci. 2003, 4, 103–112.
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic Inflammation and Disease Progression in Alzheimer Disease. Neurology 2009, 73, 768–774.
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M. Systemic Inflammation Impairs Microglial Aβ Clearance through NLRP 3 Inflammasome. EMBO J. 2019, 38, e101064.
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-Wide Association Study Identifies Variants at CLU and PICALM Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1088–1093.
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–435.
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099.
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic Variation in Aquaporin-4 Moderates the Relationship between Sleep and Brain Aβ-Amyloid Burden. Transl. Psychiatry 2018, 8, 47.
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; De Natale, E.R.; Politis, M. Alzheimer’s Disease Neuroimaging Initiative Aquaporin-4 Polymorphisms Predict Amyloid Burden and Clinical Outcome in the Alzheimer’s Disease Spectrum. Neurobiol. Aging 2021, 97, 1–9.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458.
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and Refinement of Loci Associated with Lipid Levels. Nat. Genet. 2013, 45, 1274–1283.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116.
More