Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jolien Vandewalle | + 4134 word(s) | 4134 | 2022-02-21 08:52:05 | | | |
| 2 | Bruce Ren | + 1 word(s) | 4135 | 2022-03-15 01:52:19 | | | | |
| 3 | Bruce Ren | + 1 word(s) | 4135 | 2022-03-15 01:54:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vandewalle, J. Glucocorticoid Receptor and Its Importance in (Patho)physiology. Encyclopedia. Available online: https://encyclopedia.pub/entry/20552 (accessed on 26 July 2026).
Vandewalle J. Glucocorticoid Receptor and Its Importance in (Patho)physiology. Encyclopedia. Available at: https://encyclopedia.pub/entry/20552. Accessed July 26, 2026.
Vandewalle, Jolien. "Glucocorticoid Receptor and Its Importance in (Patho)physiology" Encyclopedia, https://encyclopedia.pub/entry/20552 (accessed July 26, 2026).
Vandewalle, J. (2022, March 14). Glucocorticoid Receptor and Its Importance in (Patho)physiology. In Encyclopedia. https://encyclopedia.pub/entry/20552
Vandewalle, Jolien. "Glucocorticoid Receptor and Its Importance in (Patho)physiology." Encyclopedia. Web. 14 March, 2022.
Copy Citation
The glucocorticoid receptor (GR) is a very versatile protein that comes in several forms, interacts with many proteins and has multiple functions. Numerous therapies are based on GRs’ actions but the occurrence of side effects and reduced responses to glucocorticoids have motivated scientists to study GRs in great detail. The notion that GRs can perform functions as a monomeric protein, but also as a homodimer has raised questions about the underlying mechanisms, structural aspects of dimerization, influencing factors and biological functions
glucocorticoids
glucocorticoid receptor
dimerization
sepsis
mutants
1. Introduction
1.1. Glucocorticoids
Glucocorticoids (GCs) are steroid hormones produced by the adrenal cortex [1][2]. They have a broad range of effects including developmental, anti-inflammatory [3], metabolic [4][5] and many other functions [2]. GCs are hydrophobic molecules derived from cholesterol and hence they can easily pass cell membranes to enter in cells and exert their effects. GCs were first discovered in 1946 based on research on Addison’s disease, a rare disease, also known as primary adrenal insufficiency or hypoadrenalism [6]. The active form of GCs is known as cortisol in humans and corticosterone in rodents. Owing to their anti-inflammatory and immune-suppressive actions, and small molecular weight, GCs are among the most widely prescribed drugs worldwide and are used both in short-term and chronic settings, despite that their chronic use poses risks for side effects such as osteoporosis [7], or opportunistic infections due to the immune suppressive effects [8]. Furthermore, some patients do not respond to GC treatment and display so-called GC resistance [9], the occurrence of which varies between diseases and can also develop over the course of the treatment [9]. Over the years, much effort has been directed towards developing selective synthetic GCs with fewer side effects while keeping the therapeutic effects intact; however, success has been limited [10][11].
1.2. Glucocorticoid Receptor: Structure and Function
GCs bind the GC receptor (GR), also known as Nuclear Receptor 3 C1 (NR3C1). This is a soluble receptor belonging to the superfamily of nuclear receptors (NRs) [2][12]. It is estimated that 1000 to 2000 genes are subject to GR-mediated regulation, and some studies suggest that up to 20% of all genes are responsive to the GR. The GR regulates many pathways (e.g., gluconeogenesis [13][14], inflammatory response [3][15], fatty acid metabolism [16][17]) by regulating gene expression in various organs and tissues (e.g., liver [18][19], nervous system [20][21], adipocytes [17][22]). Over- or under stimulation of the GR results in severe phenotypes such as Cushing syndrome and Addison’s disease [23][24]. The NR family is composed of 49 members and includes other well-known receptors, such as the estrogen receptor (ER) and mineralocorticoid receptor (MR) as well as a number of orphan receptors (the ligands of which are unknown) [25]. Similar to other nuclear receptors, the protein domain configuration of the GR can be subdivided into the following four regions (Figure 1A), from N-terminus to C-terminus [25]: N-terminal domain (NTD), a DNA-binding domain (DBD), a short flexible hinge region and the ligand binding domain (LBD). The N-terminal domain is the least conserved of the four domains. It shows large differences between different nuclear receptors [25] and contains most polymorphisms in humans (mostly without deleterious effect) [26][27]. The NTD is also intrinsically disordered, making it difficult to study its structure, and no crystal structure of this region is available. However, there is some structural information known, mainly secondary structure information. The activator function domain within the NTD is an organized domain in vivo which may adopt variable conformation to induce specific responses by recruiting different cofactors based on its configuration [28]. Expression of the GR-coding gene (NR3C1) can start from several alternative transcription initiation sites, which give rise to receptor isoforms differing in the size of the NTD, each with a different transcriptional capability and tissue-specific expression [29][30]. Next to the multiple possible transcription initiation sites, the GR also has multiple possible alternative translation start sites in exon 2, leading to different translational isoforms. Finally, the GR also undergoes alternative splicing, leading to several alternative splice isoforms [31]. These isoforms are formed by splicing variations in the final introns/exons, involving the sequence coding for the LBD and sometimes the hinge region [31]. The main isoforms involve an alternative terminal splice acceptor site, resulting a different final exon used giving the GRα and GRβ isoforms [32][33]. GRα is the canonical active GR while GRβ lacks ligand binding and transactivation activities so that GRα/GRβ and GRβ/GRβ dimers cannot activate gene expression, but can bind DNA. As GRβ is always located in the nucleus, this results in an inhibition of GR activity [33][34]. Without ligand, the GR is kept in the cytoplasm in a multiprotein complex consisting of immunophilins and various chaperones in a configuration optimal for highly sensitive ligand binding [35]. When a ligand binds, the complex changes and recruits other factors [36][37], ultimately resulting in the import of GR into the nucleus through the nuclear pores, where it will exert its regulatory functions [38]. While there is one study that shows GR dimers in cytoplasm, this study was based on a strong overexpression system used in vitro [39]. Furthermore, David Bain’s group showed that GR does not form dimers spontaneously and that an additional factor, such as DNA, is needed [40].
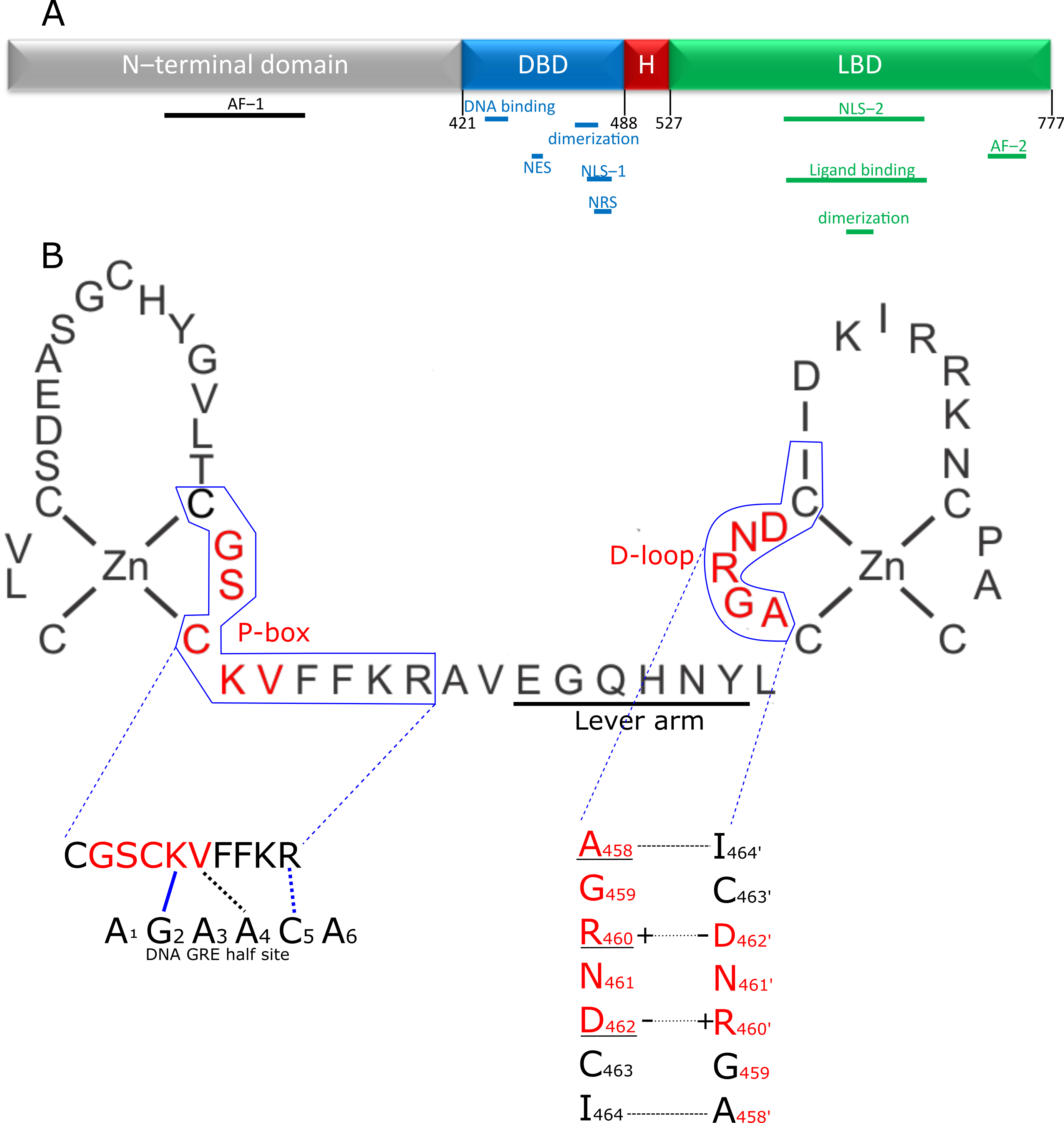
Figure 1. Structure of the GR and its DBD. (A) Domain structure of the GR. The disordered N-terminal domain contains one of the two activator functions (AF-1) for receptor transcriptional activity. The DNA binding domain (DBD; blue) contains the zinc finger element responsible for recognizing and binding the GR response element in the DNA and a second zinc finger providing the primary dimerization interface between two GR molecules. In addition, the DBD contains nuclear localization (NLS-1), nuclear retention (NRS) and nuclear export signal (NES) peptide sequences. The hinge region (H; red) is a short flexible linker connecting the DBD to the C-terminal ligand binding domain (LBD). This LBD is responsible for binding ligands in the ligand binding pocket and also contains a nuclear localization signal (NLS-2) and a second dimerization interface. Finally, the second, and most powerful, activation function (AF-2) is also located in the LBD. (B) The first zinc finger of the DBD plays a role in recognizing and binding the GRE, through the P-box. The second zinc finger is responsible for homodimerization of GR. The residues in the D-loop are especially important for this functionality. The lever arm connects both zinc fingers and changes conformation depending on the exact DNA sequence bound, transmitting DNA sequence information to the rest of the receptor. Interaction of the DBD of 1 GR partner with the DNA is depicted, with P-box shown in red. Blue lines indicate hydrogen bond interactions, black lines indicate van der Waals interactions, dotted lines indicates that the interaction is with the complementary nucleotide of the opposing strand. The K and V residues in the P-box make site specific contacts with the DNA and recognize G2 and T4′ residues respectively. The arginine outside the p-box interacts with G5′. These 3 GR-DNA interactions are of great importance, but depending on sequence context, other residues also make further stabilizing contacts with the DNA. Dimer stabilizing interaction between 2 GR molecules is depicted in the lower left figure. A458 makes a backbone-based hydrogen bond contact with I464′ (and I464 with A458′). The interface is further stabilized by 2 salt bridges formed by R460 and D462′ and D462 and R460′. Underlined residues have been subjected to mutagenesis to yield GR dimer deficient mutants, of which A458T, GRdim, has been used the most in scientific research, with many publications studying it directly (e.g., if it still forms dimers), or using it as a poorly dimerizing receptor.
It is generally accepted that GR exerts its function in the nucleus by forming a receptor homodimer and binding DNA at a specific sequence element. Initially it was thought that GR would form tetramers [41], but this was abandoned due to lack of direct evidence and the fact that support was found for the existence of dimers [42]. However, several recent studies, mainly by the group of Diego M. Presman, have once again brought the notion that GR might also exist as a tetramer, or as a dimer of dimers [43][44], to the forefront. The GR dimer model remains the most generally accepted model for GR function and GR tetramerization is an active avenue of research. Next to the dimeric and, and possible tetrameric functions of GR, the GR monomer is also thought to perform several functions. The exact functioning of GR monomers is not yet fully understood, but dimer-deficient mutants have been generated and used to infer monomeric activity. Likewise, some GR ligands that prevent dimer formation, such as compound A, can also be used. Decades ago, it was hypothesized that GR monomers would rather be transcription blockers, either by DNA binding or by inhibitory protein–protein interactions with other transcription factors, such as sequestration and tethering, while GR homodimers would be more responsible for gene upregulation via direct DNA binding. While direct monomeric DNA-binding activities have not been directly observed in a quantitative assay, there is other evidence in support (see further). Evidence for tethering and sequestration of other TFs by GR, which is believed to be a monomeric function, has been found. Since GR is supposed to bind pro-inflammatory transcription factors such as NF-kappaB as a monomer [45], and since GR likely induces expression of some genes involved in gluconeogenesis (Phosphoenolpyruvate carboxykinase 1 (PCK1) and Glucose 6-phosphate (G6PC), potentially leading to type 2 diabetes mellitus, an important side effect of GCs), as a dimer [14], this led to the paradigm that beneficial, anti-inflammatory, effects were monomer mediated and that undesirable side effects were dimer mediated. It was generally believed that skewing GR into a monomeric form or into a dimeric form could determine therapeutic outcome versus side effects profiles [46]. A balance in favor of monomers would be ideal for chronical treatment, with a minimum of side effects (from GR dimers) of long-term GR activation [47], and stronger dimerization would be better in acute settings (Figure 2) [48]. Indeed, several GR dependent gene products–thought to be dimer dependent–have been identified to have anti-inflammatory functions that can be responsible for the acute anti-inflammatory effects of GCs, as will be illustrated below.
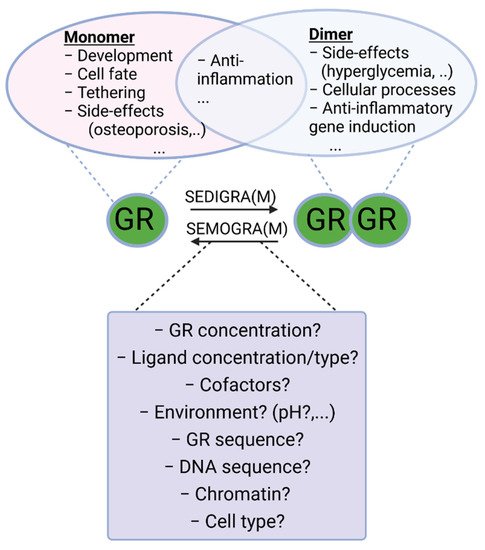
Figure 2. Monomeric and dimeric functions of GR. Work has been carried out to search for ligands that push the GR towards monomeric or dimeric action for chronic and acute inflammatory diseases, respectively. Indeed, both forms of GR possess anti-inflammatory actions trough tethering and anti-inflammatory gene induction, respectively. Most of the side-effects of GCs (hyperglycemia, glucocorticoid resistance, etc.) are ascribed to its dimeric functions, and therefore SEMOGRAMs are believed to improve the therapeutic index in chronic settings requiring long-term use of GCs. In acute inflammatory diseases, such as SIRS and sepsis, however, dimeric GR is believed to be essential to limit inflammation and therefore SEDIGRAMs are favored in these settings. A good understanding of the mechanisms determining the balance between GR monomers and dimers is needed in the search for such dissociating ligands. SEMOGRAM: selective monomerizing GR agonists and modulators, SEDIGRAM: selective dimerizing GR agonists or modulators.
Work has been carried out to search for ligands that push the GR towards monomer or dimer action as respectively Selective GR Activators and Modulators (SEGRAM), such as compound A [49], and Selective Dimerizing Glucocorticoid Receptor Agonists and Modulators (SEDIGRAM) [10]. While several effective synthetic GCs have been developed, so far this approach has not resulted in such “skewing” ligands at the bedside. Dissociating the side effects and anti-inflammatory effects of GCs solely on the basis of their monomeric or dimeric structure turned out to be unrealistic. Indeed, it has been shown that in an acute inflammatory setting, consensus GRE (dimer or higher order) gene expression is required for GC/GR effectiveness. Moreover, some side effects are not mediated via dimeric GR (see Table 1). A good understanding of the mechanisms and functions of receptor dimer formation is needed to help developing such ligands in a more educated way. The resesearchers will provide an overview of the current understanding of GR dimerization, with a focus on insights into dimerization mechanisms and the importance of intact GR dimers in pathophysiological conditions inferred from studies with mutant mice.
Table 1. Phenotypes retrieved from GRdim/dim mice in different physiological and pathological processes.
| Process | Effect in GRdim/dim Mutant | References |
|---|---|---|
| Resolution of inflammation | ||
| Antigen- and G6PI-induced arthritis | DEX protection lost | [50] |
| Serum transfer-induced arthritis | DEX protection lost | [51] |
| Contact hypersensitivity | DEX protection lost | [52] |
| PMA-induced irritative skin inflammation | DEX protection intact | [53][54] |
| Experimental autoimmune encephalomyelitis | DEX protection intact | [55] |
| Allergic airway inflammation | DEX protection lost | [56] |
| Graft- vs host disease | Increased mortality | [57] |
| TNF-induced SIRS | Increased mortality + DEX protection lost |
[58][59] |
| LPS-induced SIRS | Increased mortality + DEX protection lost |
[60][61][62][63] |
| CLP-induced septic shock | Increased mortality | [64][65] |
| Side effects | ||
| Hyperglycemia | Pred effect reduced | [66][67] |
| Osteoporosis | Pred/DEX effect intact | [68][69][70] |
| Skeletal muscle atrophy | DEX effect intact | [71] |
| Wound repair | Wound repair reduced | [72] |
| Gastroparesis and gastric acid secretion | DEX effect lost | [73] |
| Ocular hypertension leading to glaucoma | DEX effect lost | [74] |
| Glucocorticoid resistance | DEX effect lost | [75] |
| Cellular processes | ||
| Adipogenesis | No adipogenesis | [76] |
| Apoptosis | DEX effect lost | [46][77] |
| Proliferation | Proliferation reduced | [46] |
| Spatial memory | Spatial memory reduced | [78] |
| Cognitive function under stress condition | CORT effect reduced | [79] |
| Weight control | Body weight increased | [80] |
| Activation HPA axis in 6% hypoxia | Activation of HPA axis reduced | [81] |
| Trauma-induced fracture healing | Protected | [82] |
2. Role of GR Complex Formation in SIRS and Sepsis
2.1. SIRS and Sepsis
GR homodimerization is believed to be important for different physiological and pathological functions of GR, which are summarized in Table 1 (see also ref [83]). This table summarizes the findings using the GRdim/dim mice. Given the recent insights contributing to the researchers understanding of the hypersensitivity of GRdim/dim mice in systemic inflammatory response syndrome (SIRS) and sepsis, the researchers will focus the researchers discussion on this topic. The different phenotypes observed in GRdim/dim mice during acute inflammation illustrates the pleiotropic mechanisms of how dimeric GR is thought to control inflammation.
SIRS is characterized by a fast, systemic release of cytokines, such as tumor necrosis factor (TNF), interferons (IFNs), interleukin 6 (IL-6) and IL-1β, as a response to a noxious stressor such as trauma or ischemia. Sepsis evolves from an infection that causes life-threatening organ dysfunction resulting from a dysregulated host response. In contrast to SIRS, sepsis involves activation of both pro- and anti-inflammatory responses, along with abnormalities in non-immune compartments, such as the cardiovascular, metabolic and coagulation compartments [84]. According to the latest global estimates of sepsis incidence and mortality, 49 million people are affected yearly, leading to 11 million deaths, corresponding to 20% of all deaths worldwide [85]. Injection of TNF or lipopolysaccharides (LPS), the latter being cell wall components of Gram-negative bacteria, are animal models used as models for SIRS. One of the most relevant models for sepsis is the cecal ligation and puncture (CLP) model. In this model, the cecum is ligated and punctured using a needle, followed by resuscitation with antibiotic-containing fluids [86]. That GCs protect against sterile SIRS, was shown many years ago, by applying the TNF and LPS models [87][88][89].
2.2. Anti-Inflammatory Genes Induced by GR Complex Formation
GRdim/dim mice are extremely sensitive to TNF-induced SIRS [58][59]. Mortality rate is significantly higher in GRdim/dim mice compared to their WT counterparts, and this is associated with higher plasma IL-6 levels and more severe intestinal damage [58][59]. Mitogen-activated protein kinase phosphatase 1 (MKP-1) plays a key role herein. MKP-1 is induced upon dexamethasone (DEX) or TNF injection in GRwt/wt mice and not in GRdim/dim mice as a consequence of reduced binding of the mutant GR to the GRE of the MKP-1 coding gene Dusp1. Dusp1−/− mice are similarly sensitized for TNF as GRdim/dim mice and show increased levels of phosphorylated Jun N-terminal kinases (JNK), which promote apoptosis in liver tissue. Loss of Jnk2 partially rescues the TNF hypersensitivity of Dusp1–/– and GRdim/dim mice. These data illustrate the important role of GR complex formation in resisting TNF-induced SIRS through inhibiting JNK2 activation via MKP-1 activation [58]. Another anti-inflammatory gene requiring an intact GR dimerization profile is Sphingosine kinase 1 (SphK1 encoding S1P). This gene is synergistically induced by GCs and pro-inflammatory stimuli via the GR in macrophages, resulting in increased circulation of S1P during inflammation. GRdim/dim macrophages lack binding of the mutant GR to the GRE of SphK1 resulting in reduced S1P release in GRdim/dim mice upon LPS treatment. S1P is essential to limit lung inflammation induced by LPS endotoxemia as DEX can no longer protect against acute lung injury in absence of GR or SphK1 in myeloid cells, or in GRdim/dim mice [60]. A last example illustrating the importance of GR complex formation to protect against sepsis is Tsc22d3 encoding Glucocorticoid Induced Leucine Zipper (GILZ). Full expression of Tsc22d3 requires an intact GR dimerization potential [59][90]. GILZ is typically induced by GCs, but upon inflammation this gene is reduced in several cell types, such as hepatocytes and blood cells [91]. However, the mechanism behind the downregulation of GILZ in inflammatory settings is not yet understood. A possible explanation could be that due to the GC unresponsiveness in sepsis [65], the GR is unresponsive towards GR’s endogenous ligand cortisol/corticosterone, thereby leading to decreased expression of this gene. The recently registered RECORDS trial is therefore using GILZ expression in blood as a marker of corticosteroid activity for the rapid recognition of GC resistance in sepsis patients (NCT04280497). However, it should be noted that other mechanisms, next to GR-mediated transcription, can control levels of GILZ. For example, it has been illustrated that the RNA-binding protein tristetraprolin can reduce GILZ mRNA stability upon TLR activation [92]. Mice with GILZ overexpression (GILZ-tg mice) have a reduced mortality towards CLP-induced peritonitis, which could be linked to an enhanced bacterial clearance [91]. Moreover, overexpression of GILZ specifically in monocytes and macrophages similarly reduced mortality rates in the CLP model as the full GILZ-transgenic mice, and this was also associated with an increased bacterial clearance due to enhanced phagocytosis capacity of macrophages [93]. Taken together, the data suppose that anti-inflammatory genes requiring an intact GR dimerization potential (i.e., Dusp1, SphK1 and Tsc22d3) are essential to transmit the protective effects of GR in SIRS and sepsis.
2.3. Pro-Inflammatory Genes Suppressed by GR Complex Formation
In addition to its typical transactivation potential, GR dimers may downregulate hundreds of genes by interaction with IR-nGRE [94]. For example, based on studies using GRdim/dim mice, GR dimers are supposed to directly bind to IR-nGRE elements in the STAT1 promoter resulting in reduced STAT1 induction and phosphorylation. Unchallenged GRdim/dim mice, which lack the potential to bind these short DNA sequences, subsequently present a strong IFN-stimulated gene (ISG) signature. This ISG signature is gut-specific and dependent on the gut microbiota as assessed with antibiotics studies. Injection of TNF in GRdim/dim mice leads to an even more outspoken induction of these ISGs, including necroptosis master switches Ripk3, Zbp1 and Mlkl, compared to their WT counterparts [59]. The increased expression of these genes contributes to necroptotic cell death in the intestinal epithelial cells (IECs) and subsequent intestinal leakage. DEX is able to protect GRwt/wt mice challenged with a lethal TNF dose, whereas this is not the case for GRdim/dim mice challenged with their respective lethal TNF dose. The transcription factor binding motifs found in GRwt/wt-specific DEX downregulated genes were specifically ISRE and IRF elements. As such, DEX has a lower impact on the repression on TNF-induced ISGs and concomitant intestinal damage in GRdim/dim compared to GRwt/wt mice. Nec1s, a specific necroptosis inhibitor partially rescues the hypersensitivity of GRdim/dim mice to TNF, thereby illustrating the essential role for GR complex formation in resisting TNF-induced SIRS trough inhibiting necroptosis in the intestinal epithelium [59].
Next to the observed effects of the GRdim/dim mutant in IECs, this mutation also has clear effects on macrophage function. In contrast to TNF, expression of IL-1β is prolonged in GRdim/dim mice that succumb to LPS-induced shock [64]. This may be attributed to the role of GR dimerization in macrophages, as BMDMs derived from GRdim/dim mice are refractory to GC treatment as assessed by the production of IL-1β upon addition of LPS and/or DEX. Inhibiting IL-1β using an IL-1 receptor antagonist partially rescues the hypersensitivity of GRdim/dim mice to LPS, thereby illustrating an important role for complex formation in resisting LPS-induced mortality through modulating IL-1β production [64]. Interestingly, GRlysmKO mice, which lack the GR in their myeloid cells, similarly show increased susceptibility towards LPS injection, but IL-1β inhibition completely protects in this mouse model. Since GRdim/dim mice carry the point mutation in all cell types, these data suggest that GR dimerization in other cell types also contributes to survival during sepsis, and moreover, that the monomeric function of GR in myeloid cells also provides some protection [64]. Indeed, the researchers recently showed that GCs apply two key mechanisms to control endotoxemia. One at the level of macrophages, where GCs are thought to suppress TNF production in a GR monomer-dependent way, and one at the level of the intestinal epithelium, where GCs are believed to suppress TNFR1-induced ISG gene expression and necroptotic cell death in a dimer-dependent way [61]. Lastly, Osteopontin (OPN), a crucial mediator for lung inflammation, is increased in the lungs of GRdim/dim mice challenged with LPS compared to GRwt/wt mice. A partial role for GR complex formation in macrophages was found herein, as BMDMs derived from GRdim/dim mice showed a trend towards induced Opn upon LPS treatment, compared to their WT counterparts [62]. However, as this induction was not significant, a possible role for GR in other cell types contributing to Opn regulation cannot be excluded and remains to be elucidated. Collectively, these data demonstrate that suppression of pro-inflammatory genes in an assumedly GR dimer-dependent way (Stat-1, IL-1β and Opn) are essential for protection against SIRS and sepsis.
2.4. Hemodynamic and Metabolic Parameters Controlled by GR Complex Formation
Next to the direct effect of GR dimerization on the inflammatory compartment of sepsis, GR dimerization is also thought to be protective in sepsis by controlling hemodynamics and metabolic parameters. For example, GRdim/dim mice challenged with LPS have a compromised systemic hemodynamic stability and require increased norepinephrine to maintain hemodynamic stability [62]. Moreover, LPS treatment in GRdim/dim mice leads to increased levels of lactate when compared to WT mice. The higher lactate levels might be linked to a disturbed mitochondrial function in the heart of GRdim/dim animals [62]. Another clarification for the increased lactate levels of GRdim/dim mice is via reduced clearance of lactate. Indeed, the main mechanism to clear lactate is via the Cori cycle, i.e., conversion of lactate to glucose in the liver via gluconeogenesis and release of glucose in the blood which then can be taken up by peripheral tissues. Gene expression profiling of livers of prednisolone-treated GRwt/wt and GRdim/dim mice, identified reduced fold changes of typical gluconeogenic genes (e.g., Pck1, Irs1) in GRdim/dim mice compared to GRwt/wt mice, thereby suggesting a role for GR dimerization in regulating gluconeogenesis [67]. Indeed, GRdim/dim mice show more pronounced hypoglycemia after LPS challenge compared to GRwt/wt mice [95]. Interestingly, next to the role of GR dimerization in controlling glucose and lactate levels, administration of lactate leading to peak blood lactate values of 20 mM, which is the value of lactate typically observed in the sickest mice after CLP surgery, does not cause detrimental effects in GRwt/wt mice, whereas it does lead to acute lethality in GRdim/dim mice [65]. This lethality could be linked to an uncontrolled production of vascular endothelial growth factor (VEGF), resulting in vascular leakage, severe hypotension and organ damage [65]. How and where GR dimerization is supposed to control lactate toxicity remains to be studied. One possibility is through regulating lactate-induced VEGF production, for example in macrophages. Another possibility is by protecting the barrier function of the endothelium. Indeed, deletion of GR specifically in the endothelium renders mice hypersensitive for endotoxemia [96]. Whether this protective effect of GR in the endothelium is monomer- or rather dimer-dependent has not been evaluated.
Taken together, GR complex formation is important in multiple cell types (i.a., intestinal epithelium and macrophages) to protect against SIRS and sepsis (Figure 3). This protection is believed to depend on dimeric regulation of both pro- and anti-inflammatory genes through binding respectively IR-nGRE and GRE sequences. Next to their basal increased susceptibility towards SIRS and sepsis, GRdim/dim mice are also refractory to GC treatment in SIRS conditions. Moreover, GR complex formation also controls critical hemodynamic and metabolic parameters essential for surviving acute diseases such as SIRS and sepsis.
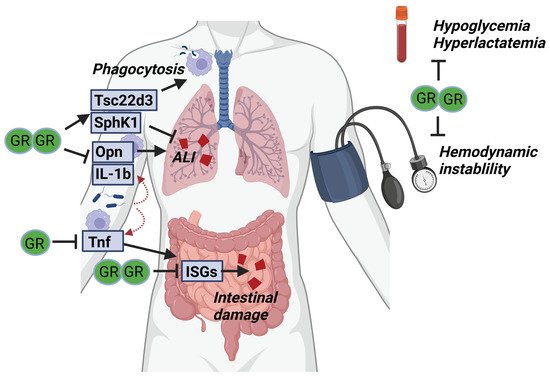
Figure 3. GR complex formation is required for surviving SIRS and sepsis. Upon infection, pro-inflammatory cytokines (Opn, IL-1b and TNF) are produced by myeloid cells and cause acute lung injury (ALI) and intestinal damage. GR dimerization is thought to be necessary to control these pro-inflammatory genes and prevent organ damage. In addition, anti-inflammatory genes (Tsc22d3, SphK1) are produced by the myeloid cells, likely in a dimer-dependent way, which is necessary to further suppress organ damage. Next to the effects of GR dimerization on the inflammatory compartment, the dimeric GR may be essential to prevent hemodynamic instability and control glucose and lactate levels during sepsis.
References
- Spiga, F.; Walker, J.J.; Terry, J.R.; Lightman, S.L. HPA axis-rhythms. Compr. Physiol. 2014, 4, 1273–1298.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545.
- Cruz-Topete, D.; Cidlowski, J. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32.
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 2007, 275, 43–61.
- Wang, J.-C.; Gray, N.E.; Kuo, T.; Harris, C.A. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2012, 2, 19.
- Ten, S.; New, M.; Maclaren, N. Clinical review 130: Addison’s disease 2001. J. Clin. Endocrinol. Metab. 2001, 86, 2909–2922.
- Buckley, L.; Humphrey, M.B. Glucocorticoid-Induced Osteoporosis. N. Engl. J. Med. 2018, 379, 2547–2556.
- Cutolo, M.; Seriolo, B.; Pizzorni, C.; Secchi, M.E.; Soldano, S.; Paolino, S.; Montagna, P.; Sulli, A. Use of glucocorticoids and risk of infections. Autoimmun. Rev. 2008, 8, 153–155.
- Wilkinson, L.; Verhoog, N.J.D.; Louw, A. Disease- and treatment-associated acquired glucocorticoid resistance. Endocr. Connect. 2018, 7, R328–R349.
- Souffriau, J.; Eggermont, M.; Van Ryckeghem, S.; Van Looveren, K.; Van Wyngene, L.; Van Hamme, E.; Vuylsteke, M.; Beyaert, R.; De Bosscher, K.; Libert, C. A screening assay for Selective Dimerizing Glucocorticoid Receptor Agonists and Modulators (SEDIGRAM) that are effective against acute inflammation. Sci. Rep. 2018, 8, 12894.
- Van Moortel, L.; Gevaert, K.; De Bosscher, K. Improved Glucocorticoid Receptor Ligands: Fantastic Beasts, but How to Find Them? Front. Endocrinol. 2020, 11, 712.
- Mazaira, G.I.; Zgajnar, N.R.; Lotufo, C.M.; Daneri-Becerra, C.; Sivils, J.C.; Soto, O.B.; Cox, M.B.; Galigniana, A.M.D. The Nuclear Receptor Field: A Historical Overview and Future Challenges. Nucl. Recept. Res. 2018, 5, 101320.
- Imai, E.; Stromstedt, P.E.; Quinn, P.G.; Carlstedt-Duke, J.; Gustafsson, J.A.; Granner, D.K. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 1990, 10, 4712–4719.
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2015; Volume 872, pp. 99–126.
- Escoter-Torres, L.; Greulich, F.; Quagliarini, F.; Wierer, M.; Uhlenhaut, N.H. Anti-inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res. 2020, 48, 8393–8407.
- Ivy, J.R.; Carter, R.N.; Zhao, J.; Buckley, C.; Urquijo, H.; Rog-Zielinska, E.A.; Panting, E.; Hrabalkova, L.; Nicholson, C.; Agnew, E.J.; et al. Glucocorticoids regulate mitochondrial fatty acid oxidation in fetal cardiomyocytes. J. Physiol. 2021, 599, 4901–4924.
- Lee, M.-J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1842, 473–481.
- Præstholm, S.M.; Correia, C.M.; Grøntved, L. Multifaceted Control of GR Signaling and Its Impact on Hepatic Transcriptional Networks and Metabolism. Front. Endocrinol. 2020, 11, 572981.
- Tronche, F.; Opherk, C.; Moriggl, R.; Kellendonk, C.; Reimann, A.; Schwake, L.; Reichardt, H.M.; Stangl, K.; Gau, D.; Hoeflich, A.; et al. Glucocorticoid receptor function in hepatocytes is essential to promote postnatal body growth. Genes Dev. 2004, 18, 492–497.
- Funder, J.W. Corticosteroid receptors and the central nervous system. J. Steroid Biochem. Mol. Biol. 1994, 49, 381–384.
- Lerch, J.K.; Madalena, K.M. Glucocorticoids and nervous system plasticity. Neural Regen. Res. 2016, 11, 37–41.
- Lee, M.-J.; Fried, S.K. The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes. Int. J. Obes. 2014, 38, 1228–1233.
- Newell-Price, J.; Bertagna, X.; Grossman, A.B.; Nieman, L.K. Cushing’s syndrome. Lancet 2006, 367, 1605–1617.
- Simpson, S.L. Addison’s disease. Br. Med. J. 1950, 2, 1164–1166.
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892.
- Kino, T. Single Nucleotide Variations of the Human GR Gene Manifested as Pathologic Mutations or Polymorphisms. Endocrinology 2018, 159, 2506–2519.
- Mackeh, R.; Marr, A.K.; Dargham, S.; Syed, N.; Fakhro, K.A.; Kino, T. Single-Nucleotide Variations of the Human Nuclear Hormone Receptor Genes in 60,000 Individuals. J. Endocr. Soc. 2017, 2, 77–90.
- Kumar, R.; Thompson, E. Folding of the glucocorticoid receptor N-terminal transactivation function: Dynamics and regulation. Mol. Cell. Endocrinol. 2012, 348, 450–456.
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, M.T.K.K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174.
- Lu, N.Z.; Cidlowski, J.A. Translational Regulatory Mechanisms Generate N-Terminal Glucocorticoid Receptor Isoforms with Unique Transcriptional Target Genes. Mol. Cell 2005, 18, 331–342.
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522.
- De Castro, M.; Elliot, S.; Kino, T.; Bamberger, C.; Karl, M.; Webster, E.; Chrousos, G.P. The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGR beta): Tissue levels, mechanism of action, and potential physiologic role. Mol. Med. 1996, 2, 597–607.
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J. Biol. Chem. 1996, 271, 9550–9559.
- Fruchter, O.; Kino, T.; Zoumakis, E.; Alesci, S.; De Martino, M.; Chrousos, G.; Hochberg, Z. The human glucocorticoid receptor (GR) isoform differentially suppresses GR-induced transactivation stimulated by synthetic glucocorticoids. J. Clin. Endocrinol. Metab. 2005, 90, 3505–3509.
- Vandevyver, S.; Dejager, L.; Libert, C. On the Trail of the Glucocorticoid Receptor: Into the Nucleus and Back. Traffic 2011, 13, 364–374.
- Czar, M.J.; Renoir, J.M.; Pratt, W.B.; Lyons, R.H.; Welsh, M.J. Evidence that the FK506-binding immunophilin heat shock protein 56 is required for trafficking of the glucocorticoid receptor from the cytoplasm to the nucleus. Mol. Endocrinol. 1995, 9, 1549–1560.
- Davies, T.H.; Ning, Y.M.; Sanchez, E.R. A new first step in activation of steroid receptors-Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 2002, 277, 4597–4600.
- Freedman, N.D.; Yamamoto, K.R. Importin 7 and importin alpha/Importin beta are nuclear import receptors for the glucocorticoid receptor. Mol. Biol. Cell 2004, 15, 2276–2286.
- Savory, J.G.A.; Preéfontaine, G.G.; Lamprecht, C.; Liao, M.; Walther, R.F.; Lefebvre, Y.A.; Hacheé, R.J.G. Glucocorticoid Receptor Homodimers and Glucocorticoid-Mineralocorticoid Receptor Heterodimers Form in the Cytoplasm through Alternative Dimerization Interfaces. Mol. Cell. Biol. 2001, 21, 781–793.
- Robblee, J.P.; Miura, M.T.; Bain, D.L. Glucocorticoid Receptor–Promoter Interactions: Energetic Dissection Suggests a Framework for the Specificity of Steroid Receptor-Mediated Gene Regulation. Biochemistry 2012, 51, 4463–4472.
- Payvar, F.; DeFranco, D.; Firestone, G.L.; Edgar, B.; Wrange, Ö.; Okret, S.; Gustafsson, J.; Yamamoto, K.R. Sequence-specific binding of glucocorticoid receptor to MTV DNA at sites within and upstream of the transcribed region. Cell 1983, 35, 381–392.
- Wrange, O.; Eriksson, P.; Perlmann, T. The Purified Activated Glucocorticoid Receptor is a Homodimer. J. Biol. Chem. 1989, 264, 5253–5259.
- Presman, D.M.; Hager, G.L. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Austin Transcr. 2017, 8, 32–39.
- Paakinaho, V.; Johnson, T.A.; Presman, D.M.; Hager, G.L. Glucocorticoid receptor quaternary structure drives chromatin occupancy and transcriptional outcome. Genome Res. 2019, 29, 1223–1234.
- Ray, A.; Prefontaine, K.E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 752–756.
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA Binding of the Glucocorticoid Receptor Is Not Essential for Survival. Cell 1998, 93, 531–541.
- De Bosscher, K.; Beck, I.M.; Ratman, D.; Berghe, W.V.; Libert, C. Activation of the Glucocorticoid Receptor in Acute Inflammation: The SEDIGRAM Concept. Trends Pharmacol. Sci. 2016, 37, 4–16.
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007.
- Lesovaya, E.; Yemelyanov, A.; Swart, A.C.; Swart, P.; Haegeman, G.; Budunova, I. Discovery of Compound A—A selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 2015, 6, 30730–30744.
- Baschant, U.; Frappart, L.; Rauchhaus, U.; Bruns, L.; Reichardt, H.M.; Kamradt, T.; Bräuer, R.; Tuckermann, J.P. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19317–19322.
- Koenen, M.; Culemann, S.; Vettorazzi, S.; Caratti, G.; Frappart, L.; Baum, W.; Krönke, G.; Baschant, U.; Tuckermann, J.P. Glucocorticoid receptor in stromal cells is essential for glucocorticoid-mediated suppression of inflammation in arthritis. Ann. Rheum. Dis. 2018, 77, 1610–1618.
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.; Stride, B.; Förster, I.; Habenicht, A.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390.
- Tuckermann, J.P.; Reichardt, H.M.; Arribas, R.; Richter, K.H.; Schütz, G.; Angel, P. The DNA Binding-Independent Function of the Glucocorticoid Receptor Mediates Repression of Ap-1–Dependent Genes in Skin. J. Cell Biol. 1999, 147, 1365–1370.
- Reichardt, H.M.; Tuckermann, J.P.; Göttlicher, M.; Vujic, M.; Weih, F.; Angel, P.; Herrlich, P.; Schütz, G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001, 20, 7168–7173.
- Schweingruber, N.; Fischer, H.J.; Fischer, L.; Brandt, J.V.D.; Karabinskaya, A.; Labi, V.; Villunger, A.; Kretzschmar, B.; Huppke, P.; Simons, M.; et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014, 127, 713–729.
- Klaßen, C.; Karabinskaya, A.; Dejager, L.; Vettorazzi, S.; Van Moorleghem, J.; Lühder, F.; Meijsing, S.H.; Tuckermann, J.P.; Bohnenberger, H.; Libert, C.; et al. Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J. Immunol. 2017, 199, 48–61.
- Baake, T.; Jörß, K.; Suennemann, J.; Roßmann, L.; Bohnenberger, H.; Tuckermann, J.P.; Reichardt, H.M.; Fischer, H.J.; Reichardt, S.D. The glucocorticoid receptor in recipient cells keeps cytokine secretion in acute graft-versus-host disease at bay. Oncotarget 2018, 9, 15437.
- Vandevyver, S.; Dejager, L.; Van Bogaert, T.; Kleyman, A.; Liu, Y.; Tuckermann, J.; Libert, C. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J. Clin. Investig. 2012, 122, 2130–2140.
- Ballegeer, M.; Van Looveren, K.; Timmermans, S.; Eggermont, M.; Vandevyver, S.; Thery, F.; Dendoncker, K.; Souffriau, J.; Vandewalle, J.; Van Wyngene, L.; et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J. Clin. Investig. 2018, 128, 3265–3279.
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klaßen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat. Commun. 2015, 6, 7796.
- Van Looveren, K.; Timmermans, S.; Vanderhaeghen, T.; Wallaeys, C.; Ballegeer, M.; Souffriau, J.; Eggermont, M.; Vandewalle, J.; Van Wyngene, L.; De Bosscher, K.; et al. Glucocorticoids limit lipopolysaccharide-induced lethal inflammation by a double control system. EMBO Rep. 2020, 21, e49762.
- Wepler, M.; Preuss, J.M.; Merz, T.; Hartmann, C.; Wachter, U.; McCook, O.; Vogt, J.; Kress, S.; Gröger, M.; Fink, M.; et al. Impaired Glucocorticoid Receptor Dimerization Aggravates LPS-Induced Circulatory and Pulmonary Dysfunction. Front. Immunol. 2020, 10, 3152.
- Silverman, M.N.; Mukhopadhyay, P.; Belyavskaya, E.; Tonelli, L.H.; Revenis, B.D.; Doran, J.H.; Ballard, B.E.; Tam, J.; Pacher, P.; Sternberg, E.M. Glucocorticoid receptor dimerization is required for proper recovery of LPS-induced inflammation, sickness behavior and metabolism in mice. Mol. Psychiatry 2012, 18, 1006–1017.
- Kleiman, A.; Hübner, S.; Parkitna, J.M.R.; Neumann, A.; Hofer, S.; Weigand, M.A.; Bauer, M.; Schmid, W.; Schütz, G.; Libert, C.; et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2011, 26, 722–729.
- Vandewalle, J.; Timmermans, S.; Paakinaho, V.; Vancraeynest, L.; Dewyse, L.; Vanderhaeghen, T.; Wallaeys, C.; Van Wyngene, L.; Van Looveren, K.; Nuyttens, L.; et al. Combined glucocorticoid resistance and hyperlactatemia contributes to lethal shock in sepsis. Cell Metab. 2021, 33, 1763–1776.e5.
- Reichardt, S.D.; Föller, M.; Rexhepaj, R.; Pathare, G.; Minnich, K.; Tuckermann, J.P.; Lang, F.; Reichardt, H.M. Glucocorticoids Enhance Intestinal Glucose Uptake Via the Dimerized Glucocorticoid Receptor in Enterocytes. Endocrinology 2012, 153, 1783–1794.
- Frijters, R.; Fleuren, W.; Toonen, E.J.; Tuckermann, J.P.; Reichardt, H.M.; van der Maaden, H.; van Elsas, A.; van Lierop, M.-J.; Dokter, W.; de Vlieg, J.; et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genom. 2010, 11, 359.
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Mandic, V.; Takacz, A.; Schmidt-Ullrich, R.; et al. Glucocorticoids Suppress Bone Formation by Attenuating Osteoblast Differentiation via the Monomeric Glucocorticoid Receptor. Cell Metab. 2010, 11, 517–531.
- Conaway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lerner, U.H. Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone 2016, 93, 43–54.
- Hachemi, Y.; Rapp, A.E.; Picke, A.-K.; Weidinger, G.; Ignatius, A.; Tuckermann, J. Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture. J. Mol. Endocrinol. 2018, 61, R75–R90.
- Waddell, D.; Baehr, L.M.; Brandt, J.V.D.; Johnsen, S.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Metab. 2008, 295, E785–E797.
- Grose, R.; Werner, S.; Kessler, D.; Tuckermann, J.; Huggel, K.; Durka, S.; Reichardt, H.M.; Werner, S. A role for endogenous glucocorticoids in wound repair. EMBO Rep. 2002, 3, 575–582.
- Reichardt, S.D.; Weinhage, T.; Rotte, A.; Föller, M.; Oppermann, M.; Lühder, F.; Tuckermann, J.P.; Lang, F.; Brandt, J.V.D.; Reichardt, H.M. Glucocorticoids Induce Gastroparesis in Mice Through Depletion of l-Arginine. Endocrinology 2014, 155, 3899–3908.
- Patel, G.C.; Millar, J.C.; Clark, A.F. Glucocorticoid Receptor Transactivation Is Required for Glucocorticoid-Induced Ocular Hypertension and Glaucoma. Investig. Opthalmol. Vis. Sci. 2019, 60, 1967–1978.
- Glantschnig, C.; Koenen, M.; Gil-Lozano, M.; Karbiener, M.; Pickrahn, I.; Williams-Dautovich, J.; Patel, R.; Cummins, C.L.; Giroud, M.; Hartleben, G.; et al. A miR-29a-driven negative feedback loop regulates peripheral glucocorticoid receptor signaling. FASEB J. 2019, 33, 5924–5941.
- Asada, M.; Rauch, A.; Shimizu, H.; Maruyama, H.; Miyaki, S.; Shibamori, M.; Kawasome, H.; Ishiyama, H.; Tuckermann, J.; Asahara, H. DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Krüppel-like factor 15 gene expression. Lab. Investig. 2010, 91, 203–215.
- Jewell, C.M.; Scoltock, A.B.; Hamel, B.L.; Yudt, M.R.; Cidlowski, J.A. Complex Human Glucocorticoid Receptor dim Mutations Define Glucocorticoid Induced Apoptotic Resistance in Bone Cells. Mol. Endocrinol. 2012, 26, 244–256.
- Oitzl, M.S.; Reichardt, H.M.; Joels, M.; de Kloet, R. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc. Natl. Acad. Sci. USA 2001, 98, 12790–12795.
- Van Looveren, K.; Van Boxelaere, M.; Callaerts-Vegh, Z.; Libert, C. Cognitive dysfunction in mice lacking proper glucocorticoid receptor dimerization. PLoS ONE 2019, 14, e0226753.
- Van Wyngene, L.; Vanderhaeghen, T.; Petta, I.; Timmermans, S.; Corbeels, K.; Van der Schueren, B.; Vandewalle, J.; Van Looveren, K.; Wallaeys, C.; Eggermont, M.; et al. ZBTB32 performs crosstalk with the glucocorticoid receptor and is crucial in glucocorticoid responses to starvation. iScience 2021, 24, 102790.
- Vanderhaeghen, T.; Timmermans, S.; Watts, D.; Paakinaho, V.; Eggermont, M.; Vandewalle, J.; Wallaeys, C.; Van Wyngene, L.; Van Looveren, K.; Nuyttens, L.; et al. Reprogramming of glucocorticoid receptor function by hypoxia. EMBO Rep. 2021, 23, e53083.
- Hachemi, Y.; Rapp, A.E.; Lee, S.; Dorn, A.K.; Krüger, B.T.; Kaiser, K.; Ignatius, A.; Tuckermann, J. Intact Glucocorticoid Receptor Dimerization Is Deleterious in Trauma-Induced Impaired Fracture Healing. Front. Immunol. 2021, 11, 3913.
- Louw, A. GR Dimerization and the Impact of GR Dimerization on GR Protein Stability and Half-Life. Front. Immunol. 2019, 10, 1693.
- Singer, M.; Deutschman, C.S.; Seymour, C.; Manu, S.-H.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock {(Sepsis-3)}. JAMA 2016, 315, 801–810.
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211.
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208.
- Bertini, R.; Bianchi, M.; Ghezzi, P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J. Exp. Med. 1988, 167, 1708–1712.
- Libert, C.; Van Bladel, S.; Brouckaert, P.; Fiers, W. The Influence of Modulating Substances on Tumor Necrosis Factor and Interleukin-6 Levels after Injection of Murine Tumor Necrosis Factor or Lipopolysaccharide in Mice. J. Immunother. 1991, 10, 227–235.
- Van Bogaert, T.; Vandevyver, S.; Dejager, L.; Van Hauwermeiren, F.; Pinheiro, I.; Petta, I.; Engblom, D.; Kleyman, A.; Schutz, G.; Tuckermann, J.; et al. Tumor necrosis factor inhibits glucocorticoid receptor function in mice: A strong signal toward lethal shock. J. Biol. Chem. 2011, 286, 26555–26567.
- Lim, H.-W.; Uhlenhaut, N.H.; Rauch, A.; Weiner, J.; Hübner, S.; Hübner, N.; Won, K.-J.; Lazar, M.A.; Tuckermann, J.; Steger, D.J. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. 2015, 25, 836–844.
- Ballegeer, M.; Vandewalle, J.; Eggermont, M.; Van Isterdael, G.; Dejager, L.; De Bus, L.; Decruyenaere, J.; Vandenbroucke, R.E.; Libert, C. Overexpression of Gilz Protects Mice Against Lethal Septic Peritonitis. Shock 2019, 52, 208–214.
- Hoppstädter, J.; Diesel, B.; Eifler, L.K.; Schmid, T.; Brüne, B.; Kiemer, A.K. Glucocorticoid-induced leucine zipper is downregulated in human alveolar macrophages upon Toll-like receptor activation. Eur. J. Immunol. 2012, 42, 1282–1293.
- Ellouze, M.; Vigouroux, L.; Tcherakian, C.; Woerther, P.; Guguin, A.; Robert, O.; Surenaud, M.; Tran, T.; Calmette, J.; Barbin, T.; et al. Overexpression of GILZ in macrophages limits systemic inflammation while increasing bacterial clearance in sepsis in mice. Eur. J. Immunol. 2019, 50, 589–602.
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread Negative Response Elements Mediate Direct Repression by Agonist- Liganded Glucocorticoid Receptor. Cell 2011, 145, 224–241.
- Wepler, M.; Preuss, J.M.; Merz, T.; McCook, O.; Radermacher, P.; Tuckermann, J.P.; Vettorazzi, S. Impact of downstream effects of glucocorticoid receptor dysfunction on organ function in critical illness-associated systemic inflammation. Intensiv. Care Med. Exp. 2020, 8, 37.
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311.
More
Information
Subjects:
Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
884
Revisions:
3 times
(View History)
Update Date:
15 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No