 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Masatoshi Jinnin | + 2454 word(s) | 2454 | 2022-03-03 04:13:09 | | | |
| 2 | Catherine Yang | Meta information modification | 2454 | 2022-03-11 05:19:16 | | |
Video Upload Options
Vascular anomalies include various diseases, which are classified into two types according to the International Society for the Study of Vascular Anomalies (ISSVA) classification: vascular tumors with proliferative changes of endothelial cells, and vascular malformations primarily consisting of structural vascular abnormalities. Vascular lesions had been habitually termed ‘hemangioma’ or ‘angioma’ in both Japan and Europe/the United States based on the impression that most anomalies are ‘tumors’. On the contrary, hemangioma simplex and cavernous hemangioma, for example, are actually morphological abnormalities of capillary blood vessels or veins, respectively, despite the disease name “hemangioma”. These diseases differ from tumors in a narrow sense, which refers to autonomous cell proliferation. In addition to such problems with disease naming and nomenclature, vascular lesions can occur at various ages and in various organs, meaning they may require treatment in various hospital departments, so improved common terms/language are essential for mutual understanding.
1. Venous Malformation
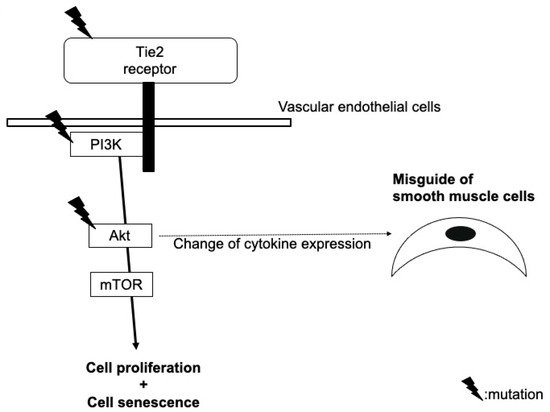
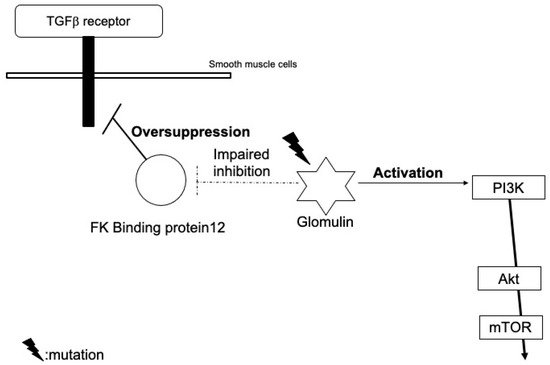
2. Glomuvenous Malformation
3. Lymphatic Malformation
4. Arteriovenous Malformation
5. Klippel–Trenaunay Syndrome
6. Sturge–Weber Syndrome
7. Infantile Hemangioma
8. Tufted Angioma and Kaposiform Hemangioendothelioma
References
- Vikkula, M.; Boon, L.M.; Carraway, K.L., 3rd; Calvert, J.T.; Diamonti, A.J.; Goumnerov, B.; Pasyk, K.A.; Marchuk, D.A.; Warman, M.L.; Cantley, L.C.; et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell 1996, 87, 1181–1190.
- Limaye, N.; Wouters, V.; Uebelhoer, M.; Tuominen, M.; Wirkkala, R.; Mulliken, J.B.; Eklund, L.; Boon, L.M.; Vikkula, M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 2009, 41, 118–124.
- Brouillard, P.; Ghassibé, M.; Penington, A.; Boon, L.M.; Dompmartin, A.; Temple, I.K.; Cordisco, M.; Adams, D.; Piette, F.; Harper, J.I.; et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): Evidence for a founder effect. J. Med. Genet. 2005, 42, e13.
- Fereydooni, A.; Dardik, A.; Nassiri, N. Molecular changes associated with vascular malformations. J. Vasc. Surg. 2019, 70, 314–326.e1.
- Castillo, S.D.; Tzouanacou, E.; Zaw-Thin, M.; Berenjeno, I.M.; Parker, V.E.; Chivite, I.; Mila-Guasch, M.; Pearce, W.; Solomon, I.; Angulo-Urarte, A.; et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci. Transl. Med. 2016, 8, 332ra43.
- Castillo, S.D.; Baselga, E.; Graupera, M. PIK3CA mutations in vascular malformations. Curr. Opin. Hematol. 2019, 26, 170–178.
- Blesinger, H.; Kaulfuss, S.; Aung, T.; Schwoch, S.; Prantl, L.; Rossler, J.; Wilting, J.; Becker, J. PIK3CA mutations are specifically localized to lymphatic endothelial cells of lymphatic malformations. PLoS ONE 2018, 13, e0200343.
- Couto, J.A.; Huang, A.Y.; Konczyk, D.J.; Goss, J.A.; Fishman, S.J.; Mulliken, J.B.; Warman, M.L.; Greene, A.K. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am. J. Hum. Genet. 2017, 100, 546–554.
- Palmieri, M.; Curro, A.; Tommasi, A.; Di Sarno, L.; Doddato, G.; Baldassarri, M.; Frullanti, E.; Giliberti, A.R.; Fallerini, C.; Spinazzola, A.; et al. Cell-free DNA next-generation sequencing liquid biopsy as a new revolutionary approach for arteriovenous malformation. JVS Vasc. Sci. 2020, 1, 176–180.
- Dekeuleneer, V.; Seront, E.; Van Damme, A.; Boon, L.M.; Vikkula, M. Theranostic advances in vascular malformations. J. Investig. Dermatol. 2020, 140, 756–763.
- Smits, P.J.; Konczyk, D.J.; Sudduth, C.L.; Goss, J.A.; Greene, A.K. Endothelial MAP2K1 mutations in arteriovenous malformation activate the RAS/MAPK pathway. Biochem. Biophys. Res. Commun. 2020, 529, 450–454.
- Fish, J.E.; Flores Suarez, C.P.; Boudreau, E.; Herman, A.M.; Gutierrez, M.C.; Gustafson, D.; DiStefano, P.V.; Cui, M.; Chen, Z.; De Ruiz, K.B.; et al. Somatic gain of KRAS function in the endothelium is sufficient to cause vascular malformations that require MEK but not PI3K signaling. Circ. Res. 2020, 127, 727–743.
- Castel, P.; Carmona, F.J.; Grego-Bessa, J.; Berger, M.F.; Viale, A.; Anderson, K.V.; Bague, S.; Scaltriti, M.; Antonescu, C.R.; Baselga, E.; et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci. Transl. Med. 2016, 8, 332ra42.
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979.
- Nguyen, V.; Hochman, M.; Mihm, M.C., Jr.; Nelson, J.S.; Tan, W. The pathogenesis of port wine stain and sturge weber syndrome: Complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int. J. Mol. Sci. 2019, 20, 2243.
- Asadi, S. The role of genetic mutations in gene GNAQ in Sturge-Weber syndrome. J. Cell Signal Damage Assoc. Mol. Patterns 2020, 1, 15–19.
- Ferrara, N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS 2005, 94, 209–231.
- Boye, E.; Yu, Y.; Paranya, G.; Mulliken, J.B.; Olsen, B.R.; Bischoff, J. Clonality and altered behavior of endothelial cells from hemangiomas. J. Clin. Investig. 2001, 107, 745–752.
- Khan, Z.A.; Melero-Martin, J.M.; Wu, X.; Paruchuri, S.; Boscolo, E.; Mulliken, J.B.; Bischoff, J. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood 2006, 108, 915–921.
- Kleinman, M.E.; Tepper, O.M.; Capla, J.M.; Bhatt, K.A.; Ceradini, D.J.; Galiano, R.D.; Blei, F.; Levine, J.P.; Gurtner, G.C. Increased circulating AC133+ CD34+ endothelial progenitor cells in children with hemangioma. Lymphat. Res. Biol. 2003, 1, 301–307.
- Yu, Y.; Fuhr, J.; Boye, E.; Gyorffy, S.; Soker, S.; Atala, A.; Mulliken, J.B.; Bischoff, J. Mesenchymal stem cells and adipogenesis in hemangioma involution. Stem Cells 2006, 24, 1605–1612.
- Khan, Z.A.; Boscolo, E.; Picard, A.; Psutka, S.; Melero-Martin, J.M.; Bartch, T.C.; Mulliken, J.B.; Bischoff, J. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J. Clin. Investig. 2008, 118, 2592–2599.
- Ritter, M.R.; Dorrell, M.I.; Edmonds, J.; Friedlander, S.F.; Friedlander, M. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc. Natl. Acad. Sci. USA 2002, 99, 7455–7460.
- Melero-Martin, J.M.; De Obaldia, M.E.; Kang, S.Y.; Khan, Z.A.; Yuan, L.; Oettgen, P.; Bischoff, J. Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ. Res. 2008, 103, 194–202.
- St Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202.
- Hashimoto, A.; Kunimoto, K.; Kawaguchi, A.; Inaba, Y.; Kaminaka, C.; Yamamoto, Y.; Kakimoto, N.; Suenaga, T.; Takeuchi, T.; Suzuki, H.; et al. Analysis of onset and clinical characteristics in Japanese patients with infantile hemangioma. Drug Discov. Ther. 2021, 15, 210–213.
- Lim, Y.H.; Fraile, C.; Antaya, R.J.; Choate, K.A. Tufted angioma with associated Kasabach-Merritt phenomenon caused by somatic mutation in GNA14. Pediatr. Dermatol. 2019, 36, 963–964.
- Croteau, S.E.; Liang, M.G.; Kozakewich, H.P.; Alomari, A.I.; Fishman, S.J.; Mulliken, J.B.; Trenor, C.C., 3rd. Kaposiform hemangioendothelioma: Atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J. Pediatr. 2013, 162, 142–147.
- Lewis, D.; Vaidya, R. Kasabach merritt syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Lim, Y.H.; Bacchiocchi, A.; Qiu, J.; Straub, R.; Bruckner, A.; Bercovitch, L.; Narayan, D.; Yale Center for Mendelian Genomics; McNiff, J.; Ko, C.; et al. GNA14 somatic mutation causes congenital and sporadic vascular tumors by MAPK activation. Am. J. Hum. Genet. 2016, 99, 443–450.
- Ten Broek, R.W.; Koelsche, C.; Eijkelenboom, A.; Mentzel, T.; Creytens, D.; Vokuhl, C.; van Gorp, J.M.; Versleijen-Jonkers, Y.M.; van der Vleuten, C.J.; Kemmeren, P.; et al. Kaposiform hemangioendothelioma and tufted angioma-(epi)genetic analysis including genome-wide methylation profiling. Ann. Diagn. Pathol. 2020, 44, 151434.

