Vascular anomalies include various diseases, which are classified into two types according to the International Society for the Study of Vascular Anomalies (ISSVA) classification: vascular tumors with proliferative changes of endothelial cells, and vascular malformations primarily consisting of structural vascular abnormalities. Vascular lesions had been habitually termed ‘hemangioma’ or ‘angioma’ in both Japan and Europe/the United States based on the impression that most anomalies are ‘tumors’. On the contrary, hemangioma simplex and cavernous hemangioma, for example, are actually morphological abnormalities of capillary blood vessels or veins, respectively, despite the disease name “hemangioma”. These diseases differ from tumors in a narrow sense, which refers to autonomous cell proliferation. In addition to such problems with disease naming and nomenclature, vascular lesions can occur at various ages and in various organs, meaning they may require treatment in various hospital departments, so improved common terms/language are essential for mutual understanding.
1. Venous Malformation
Venous malformation was previously known as “cavernous hemangioma”. It manifests as a soft subcutaneous mass with a normal to blue-purple coloring surface. Many cases are sporadic, while hereditary types are known as “familial mucocutaneous venous malformations”.
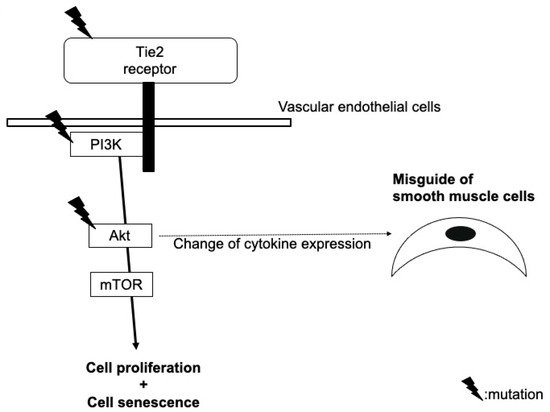
Among vascular anomalies, venous malformations are one of the diseases in which causative genes are primarily identified. A genetic analysis using blood samples from patients with familial mucocutaneous venous malformations initially identified the presence of heterozygous germline missense point mutations: a C-to-T nucleotide transition leading to an arginine-to-tryptophan substitution at position 849 in the kinase domain of Tie2
[1][2]. It was unclear at that time, however, why localized lesions occurred without involving all blood vessels, despite the presence of germline mutations in all cells of a patients’ body, and why the penetrance of the mutation was low.
Other several somatic heterozygous mutations (e.g., p.L914F) were, thereafter, identified in a Tie2 gene allele, which was normal in blood DNA and in the lesional DNA of patients with familial venous malformations
[2][3]. Such ‘second-hit’ mutations in lesional DNA explain the reasons for localized lesions with the noninvolvement of all blood vessels and the low penetrance of the mutation. Tie2 mutations are now known to also be found in patients with sporadic venous malformations, and it is recognized that some patients also have mutations in PIK3CA or Akt, which are downstream molecules of Tie2. The mechanism by which these gene mutations induce characteristic morphological abnormalities of veins still requires clarification, but there are several hypotheses (
Figure 12).
Figure 12.
Hypothetical model of the role of mutations in Tie2, PIK3CA, and Akt in the pathogenesis of venous malformation.
2. Glomuvenous Malformation
In the ISSVA classification, a glomuvenous malformation is classified into the category of venous malformations. Histologically, the lesions are due to the proliferation of immature smooth muscle cells (glomus cells). Sporadic cases can be painful and appear below the nails, while hereditary cases can manifest as multiple lesions.
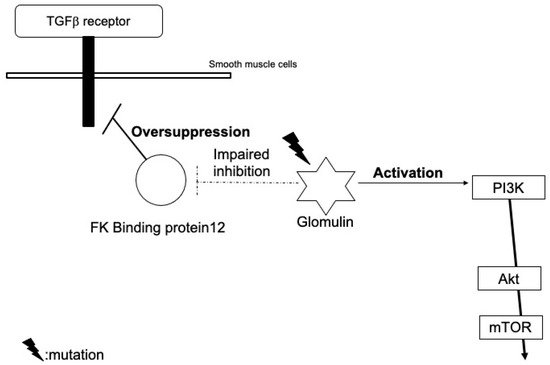
Heterozygous germline mutations in the glomulin gene have been identified in the gene analysis of blood DNA in patients with familial glomuvenous malformations (e.g., the deletion mutation of 157delAAGAA and point mutation c.C108A), but with a low penetrance. An analysis of the lesional DNA in individual patients confirmed second-hit mutations of the glomulin gene (980delCAGAA)
[3][7].
This somatic mutation is not likely to affect the expression, but cause the loss-of-function of glomulin. Wild-type glomulin proteins bind to and inhibit the FK binding protein 12 (
Figure 23). The FK binding protein also blocks TGF-β signals, but mutant glomulin cannot inhibit the FK binding protein, which leads to an excessive reduction in TGF-β signals
[3][7]. Immature glomus cells may proliferate due to the impairment of the TGF-β-mediated smooth muscle cell differentiation.
Figure 23.
Hypothetical model of the role of glomulin mutations in the pathogenesis of glomuvenous malformation.
Furthermore, they may also activate PI3K signals through interactions with the hepatocyte growth factor receptor, c-met
[4][8].
3. Lymphatic Malformation
Lymphatic malformation is a congenital lymphatic dysplasia. It is classified into two types: the macrocystic type, in which 1–3 large cystic lesions are present, and the microcystic type, in which small cystic lesions are aggregated. In addition to an abnormal expression of the molecules involved in lymphangiogenesis, such as the vascular endothelial growth factor (VEGF)-C and VEGF receptor type3 (VEGFR3), PIK3CA mutations have been detected in lymphatic ECs of the lesions
[5][4]. Similar to epithelial cancers, the most common PIK3CA mutations found in lymphatic malformation are activating mutations in the helical (p.E542K) and kinase (p.H1047R and p.H1047L) domains
[6][9]. p.E109del, p.C420R, p.E545K, p.Q546K, and p.H1047L have also been reported
[7][10].
The early embryonic activation of p110α was indicated in a mouse model study to, predominantly, lead to the formation of lesions that recapitulated features of human macrocystic lymphatic malformation, whereas its activation during late embryonic or neonatal development resulted in microcystic lesions. Accordingly, the cell circumstances/environments at the timing of gene mutation occurrence may explain differences in the clinical features of lymphatic malformations.
4. Arteriovenous Malformation
Arteriovenous malformation is defined as a congenital abnormal connection of arteries and veins, which disrupts normal blood flow and oxygen circulation. According to the Schobinger Staging system, clinical manifestation varies from cutaneous warm, pink-blue shunting to ulceration, bleeding, and cardiac failure.
Mutations in the genes of the RAS pathway, including KRAS (e.g., p.G12D and p.G12V), MAP2K1 (e.g., p.F53L, p.Q56P, p.K57N, p.Q58del, and p.D67Y), and BRAF (e.g., p.V600E), have been detected
[8][9][10][11,12,13]. Although the influence of these mutations on the expression and activation of RAS pathway molecules is not fully understood, downstream MEK/ERK signals have been found to be activated in the lesion
[11][14]. In addition, morphological changes in ECs, an increase in sprouting behavior, the enlargement of the vessel lumen, and abnormal connections between arteries and veins without cell proliferation are induced by RAS activation; gene mutations in the RAS pathway may be, therefore, strongly involved in the pathogenesis
[12][15].
5. Klippel–Trenaunay Syndrome
Klippel–Trenaunay–Weber syndrome was previously defined as a triad of hemangioma simplex, varicose veins, and bony/soft tissue hypertrophy involving an extremity. It is currently classified into two distinct diseases; Parkes Weber syndrome is defined as vascular malformations involving high-flow arterial components (arteriovenous fistulae) in addition to affected-limb hypertrophy, whereas Klippel–Trenaunay syndrome is combination of slow-flow vascular malformations (capillary, lymphatic, and venous components) accompanied with limb hypertrophy.
PIK3CA mutations, on the other hand, are also detected in lesions of venous or lymphatic malformations. Germline PIK3CA mutations, as described above, lead to embryonic lethality. Venous or lymphatic malformations are usually localized, so mutations are thought to occur late during development, affecting a single clone of ECs
[5][4]. On the other hand, mosaic mutations in the early embryonic phase may result in PROS. These PIK3CA mutations have also been detected in adult epithelial tumors, such as breast cancers or colon cancers, but other gene mutations are also present in many cases
[5][4]. However, PIK3CA mutations alone do not induce overgrowth in mice models
[13][5], so there is also a possibility that environmental factors, other gene mutations, or mutations in other cells, such as fibroblasts, may be required for the formation of overgrowth lesions
[5][13][4,5].
6. Sturge–Weber Syndrome
This syndrome was originally regarded as neurocutaneous disease, which involves a facial hemangioma simplex reaching the first branch of the trigeminal nerve, ophthalmologic abnormalities (especially congenital glaucoma), and neurologic signs (seizure, mental retardation, hemiparesis).
Mosaic gene mutations of GNAQ (e.g., p.R183Q) or GNA11 (p.R183C, p.R183H, p.Q209L, and p.Q209P) in ECs were recently detected
[14][15][16][17,18,19]. They did not change the protein expression, and are thought to activate downstream MEK/ERK pathways, but still require clarification of the detailed mechanism. Furthermore, the mechanism by which the mutated GNAQ/GNA11 genes induce capillary malformations is also still unknown, but there are several hypotheses.
7. Infantile Hemangioma
Concerning vascular tumors, causative genes have not been sufficiently identified in comparison to vascular malformations. For example, infantile hemangioma is a benign vascular tumor caused by the uncontrolled proliferation of vascular ECs. Causative genes have not been described in the ISSVA classification 2018, but cultured infantile hemangioma-derived ECs (hemECs) have been found to have an increased proliferation and migration activity
[17][21]. Furthermore, they are clonal based on analyses of X-chromosome inactivation, while non-ECs from the lesions did not exhibit clonality
[18][22]. Infantile hemangioma has, therefore, been thought to be caused by an “intrinsic abnormal activation” of ECs that leads to local clonal expansion, rather than a secondary response of ECs to external factors.
According to another hypothesis, tumor cells of infantile hemangioma might be derived from undifferentiated stem cells or progenitor cells (e.g., dormant angioblasts or cells recruited to the lesions from a reservoir of stem/progenitor cells). Khan et al. identified infantile hemangioma-derived endothelial progenitor cells (hemEPCs) by their mRNA expression patterns or their response to endostatin, and indicated that hemEPCs and hemECs share common properties with cord blood EPCs
[19][25]. In addition, Kleinman reported an elevation of the level of circulating EPCs in babies with infantile hemangioma, and that they may be recruited into tumors at the proliferating stage, leading to tumor formation
[20][26]. Furthermore, mesenchymal stem cells derived from infantile hemangioma (hemMSCs) at the proliferating stage exhibited higher adipogenic activity than those from lesions at the involuting stage and from normal skin, supporting the hypothesis that MSCs reside in the tumor and are the source of fibrofatty tissues seen in the involuted phase
[21][27]. Infantile hemangioma-derived stem cells (hemSCs) express CD90, a mesenchymal cell marker, which is one of the genes upregulated in proliferating lesions compared to involuting lesions
[22][23][28,29]. Normal human dermal microvascular ECs (HDMECs) required mesenchymal supporting cells to form vessels in subcutaneous implants in immunodeficient mice
[24][30], whereas the implantation of hemSCs alone could form functional vessels exhibiting an infantile hemangioma-like phenotype with a high GLUT-1 expression and could differentiate into adipocytes
[22][28]. Other cell types, including hemECs, hemEPCs, cord blood EPCs, normal human fibroblasts, and bone marrow-derived mesenchymal stem cells, did not form vessels in the same mouse model. Accordingly, hemSC may mainly represent progenitor cells for infantile hemangioma.
As the mechanism of VEGFR1 downregulation, we identified a putative binding site of the nuclear factor in activated T cells (NFAT) on a region of the VEGFR1 promoter. NFAT is known to be activated by the Ca++/calmodulin-dependent phosphatase calcineurin, and a low NFATc2 activation and low basal levels of cytoplasmic Ca++ were found in hemECs. In addition, the expression of active β1 integrin determined by the HUTS-21 antibody was reduced in hemECs compared with in HDMECs. A link between integrins and Ca++ signaling has been well established, and the inactivation of the β1 integrin may cause the suppression of the Ca++-NFAT-VEGFR1 pathway in hemECs. Consistently, the activation of the β1 integrin by its stimulatory antibody in hemECs could reduce VEGFR2 phosphorylation, induce the binding of NFATc2 to the VEGFR1 promoter, and stimulate the expression of VEGFR1.
TEM8 is also known as Anthrax toxin receptor 1, and is an integrin-like receptor expressed in ECs
[25][34]. The overexpression of wild-type TEM8 in hemECs with mutated TEM8 induced the amount of activated β1 integrin, stimulated the association between NFATc2 and the Flt-1 promoter, and upregulated VEGFR1 expression. In turn, the overexpression of mutant TEM8 in HDMECs induced an infantile hemangioma-like phenotype. The overexpression of wild-type VEGFR2 in hemECs with mutated VEGFR2 also normalized the infantile hemangioma-like phenotype.
Immunoprecipitation assays showed that VEGFR2 could form complexes with β1 integrin and TEM8 in hemECs, and that mutations in VEGFR2 and TEM8 resulted in an increased interaction among the three proteins. An increased complex formation among the three molecules and the subsequent inactivation of the β1 integrin-NFATc2-VEGFR1 pathway are features that are common to all hemECs that have been tested, even to hemECs in which we did not yet find mutations.
However, because hemECs exhibit clonality, the germline mutations in TEM8 and VEGFR2 must be associated with a secondary somatic event to trigger the clonal expansion of tumor cells within the hemangioma lesions
[18][22]. We, therefore, conclude that the changes to the TEM8 and VEGFR2 amino acid sequence represent risk factor mutations for infantile hemangioma, similar to familial cases of venous malformation or glomuvenous malformations as described above. Given their emergence after birth and the age-dependent involution of infantile hemangioma, we also hypothesized that physiological events, including perinatal hypoxia or mechanical stress, during delivery would be a trigger of hemangioma formation in infants with germline mutations with TEM8 and VEGFR2.
On the other hand, based on above hypothesis, we also tried to confirm that head and neck lesions were more frequent in the group in which infantile hemangioma appeared after birth compared with the patients in which it was present at birth
[26][36]. A slight such tendency was observed, but the difference was not statistically significant. Meanwhile, we unexpectedly found a statistically significant increase in the frequency of multiple lesions in infantile hemangioma after birth compared with those present at birth. This may indicate there may be different triggers between the two groups. In other words, infantile hemangioma present at birth are likely caused by a local trigger, while lesions appearing after birth may be induced by systemic factors in addition to local triggers, such as cytokines related to systemic neovascularization after birth.
8. Tufted Angioma and Kaposiform Hemangioendothelioma
TA is a relatively rare and benign vascular tumor of infancy clinically presenting as violaceous, indurated, or nodular plaques with pain, focal hyperhidrosis, and hypertrichosis. Histopathologically, the tumor shows confluent lobules or “cannonballs” of spindled ECs with slitlike lumina embedded in a fibrotic background
[27][37]. Some lesions show spontaneous regression, but the underlying pathobiology of TA is poorly understood.
On the other hand, KHE, first reported by Zukerberg et al. in 1993, is characterized by a locally aggressive/borderline malignant behavior and morphological features similar to Kaposi sarcoma. TA is currently thought to be the dermal counterpart of KHE with a benign clinical course due to the superficial origin.
Kasabach–Merritt syndrome (KMS), a consumptive coagulopathy characterized by profound thrombocytopenia, hypofibrinogenemia, and microangiopathic anemia with up to 24 percent mortality
[27][28][37,38], may affect approximately 70% of all patients with KHE and approximately 10% of patients with TA
[29][39].
Although the etiologies of TA, KHE, and KMP are poorly understood, TA and KHE mainly occur in early childhood, suggesting the involvement of gene mutations in their pathogenesis. Lim et al. found
GNA14 mutations c.614A>T (p.Q205L) in these lesions
[30][40]. However, these mutations are not specific to TA and KHE, and they were also detected in other rare vascular tumors, which are potential differential diagnoses
[27][28][37,38]. Ten Broek et al. analyzed the epigenetic genomewide methylation profile, and demonstrated that KHE and TA share a common origin without a relationship to vascular malformation
[31][41].