Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marijana Radić Stojković | + 4629 word(s) | 4629 | 2022-03-03 10:57:27 | | | |
| 2 | Marijana Radić Stojković | Meta information modification | 4629 | 2022-03-10 11:35:37 | | | | |
| 3 | Marijana Radić Stojković | -31 word(s) | 4598 | 2022-03-10 11:40:13 | | | | |
| 4 | Rita Xu | -452 word(s) | 4146 | 2022-03-11 03:07:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Radić Stojković, M. ATT Triplex and DNA. Encyclopedia. Available online: https://encyclopedia.pub/entry/20423 (accessed on 24 June 2026).
Radić Stojković M. ATT Triplex and DNA. Encyclopedia. Available at: https://encyclopedia.pub/entry/20423. Accessed June 24, 2026.
Radić Stojković, Marijana. "ATT Triplex and DNA" Encyclopedia, https://encyclopedia.pub/entry/20423 (accessed June 24, 2026).
Radić Stojković, M. (2022, March 10). ATT Triplex and DNA. In Encyclopedia. https://encyclopedia.pub/entry/20423
Radić Stojković, Marijana. "ATT Triplex and DNA." Encyclopedia. Web. 10 March, 2022.
Copy Citation
Due to the involvement of DNA:RNA hybrids and triplex helices in many essential functions in cells, this entry’s main aim is to detect benzothiazole based moieties with selective binding or spectroscopic response to these nucleic structures compared to regular (non-hybrid) DNA and RNA duplexes and single-stranded forms.
benzothiazoles
competition dialysis
DNA:RNA hybrids
ATT triplex
RNase H
circular dichroism spectroscopy
1. Introduction
Nucleic acids are molecular targets for many drugs in cancer therapy due to their essential functions in cells (replication, transcriptional and translational regulation, and enzymatic reactions) [1]. Nucleic acid structures present a wide variety of shapes with varying major and minor groove widths that can be recognized by small molecules using a non-specific (mainly electrostatic) binding along the nucleic acid exterior, a specific groove binding, and intercalation (insertion of planar aromatic molecules between base pairs) [2].
Many studies have been directed towards the rational design of small molecules that will selectively recognize multistranded structures of nucleic acids, such as triplexes [2][3][4][5][6][7].
In most cases, triplexes in solution contain conformational features that are intermediate between A- and B-form. The parallel- or pyrimidine-motif (Py) has a C- or T-rich third strand bound in a parallel orientation to the duplex homopurine strand, while the antiparallel- or purine-motif (Pu) has the opposite orientation and a primarily A- or G-rich third strand [8][9].
Specific ligands can stabilize triple helices through intercalation. For example, it has been demonstrated that ethidium bromide stabilizes polydA–2polydT (ATT) with T⋅A × T triplets, while benzopyridoindole derivatives can stabilize triple helices containing both T⋅A × T and C⋅G × C+ base triplets [3]. The targeting of triplexes has recently been the focus of the antigene strategy for gene regulation. [10] The ability to target specific genes to modulate their structure and/or function in the genome has far-reaching implications in biology, biotechnology, and medicine (Figure 1) [11][12][13][14][15].
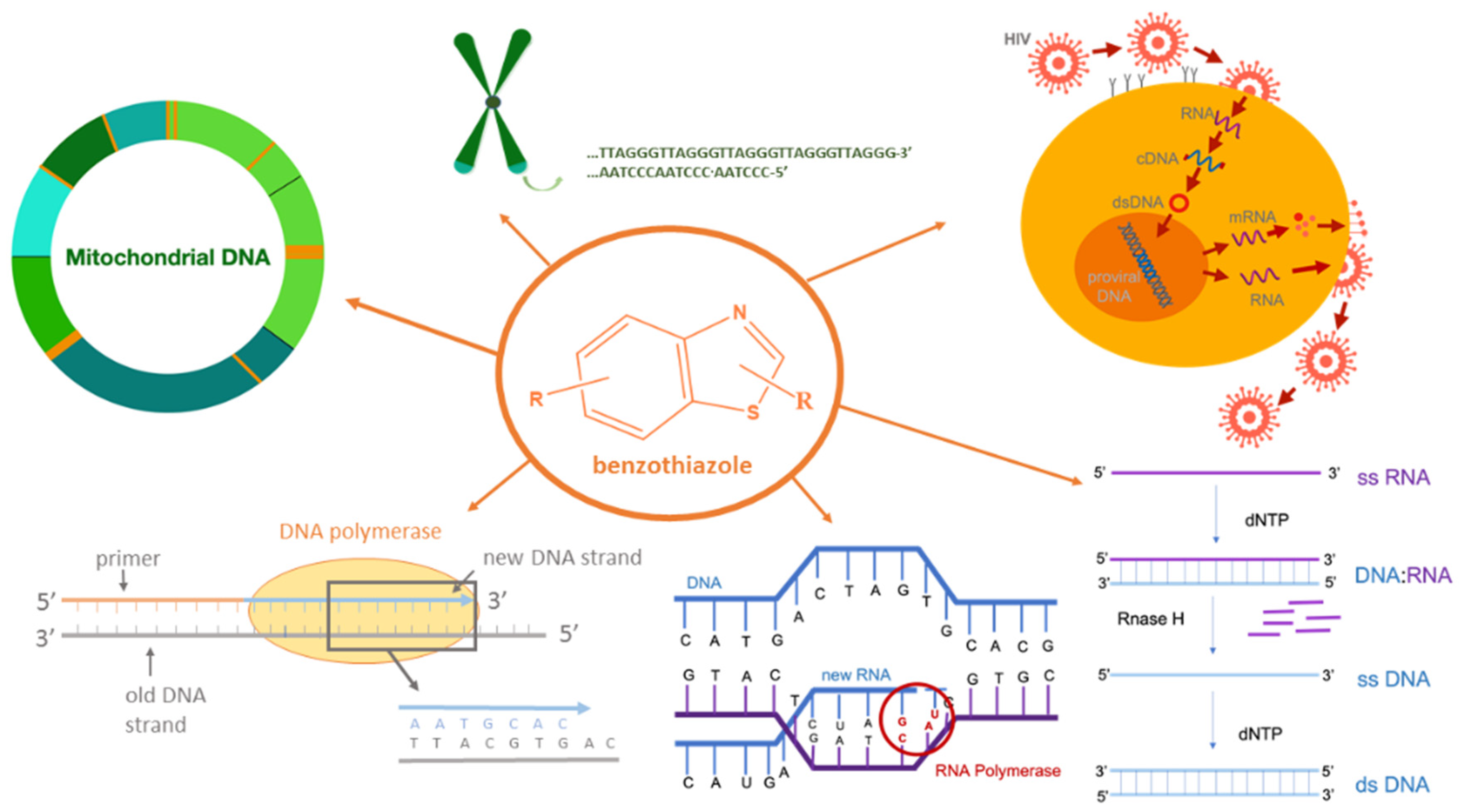
Figure 1. Potential biological activities of benzothiazole compounds.
DNA:RNA hybrids are formed as intermediate structures during many biologically important processes, such as DNA replication, transcription, and telomere replication and replication of HIV by reverse transcription (Figure 1) [16][17][18][19][20]. They can also form R-loops that have been detected in various organisms from bacteria to mammals and play crucial roles in regulating gene expression, DNA and histone modifications, immunoglobulin class switch recombination, DNA replication, and genome stability [21]. Small organic molecules with the ability to selectively inhibit DNA replication via Okazaki fragments, thus also blocking transcription, have a great potential in treating cancer because the replication is often accelerated in cancer cells (Figure 1). In addition, small molecules selective for hybrid duplexes have potential therapeutic applications as telomerase and RNaseH inhibitors [22][23][24][25].
There are few examples in the literature dedicated to the discovery of compounds that selectively bind to DNA:RNA hybrids [26][27][28]. Literature sources point to the existence of a small number of ligands with selective binding to DNA:RNA hybrids [26][27][29][30][31].
In thorough studies of Arya and Chaires, a common structural motif that preferentially binds to the hybrid structures was identified employing rapid screening assays, the competition dialysis, and thermal denaturation of mixtures [26][27][29][31][32][33]. Several compounds containing the common motif-planar aromatic ring system with a “bay” region, such as ethidium bromide, coralyne, aminoglycoside, propidium, thiazole orange and ellipticine, demonstrated preferential binding to hybrid duplexes, among other nucleic acid structures.
As the benzothiazole structure also meets this criterion of the common motif, researchers have chosen for this study nine benzothiazole derivatives, synthesized by Racane et al., [34][35][36], which demonstrated high antiproliferative activity on a panel of cancer cell lines.
2. Characterization of Compounds in Aqueous Medium
This study included nine cationic compounds: bis-benzothiazolyl-pyridines (1–3, group 1), 2-thienyl/2-benzothienyl-substituted 6-(2-imidazolinyl)benzothiazoles (4–6, group 2), and 2-aryl/heteroaryl-substituted 6-(2-imidazolinyl)benzothiazoles (7–9, group 3) (Scheme 1). All compounds were soluble (c = 5 × 10−3 mol dm−3) in redistilled water or aqueous buffer (sodium cacodylate/HCl buffer, I = 0.05 mol dm−3). Buffered solutions of studied compounds were stable for more days. The absorbancies of studied compounds were proportional to their concentrations up to c = 2 × 10−5 mol dm−3. Linear changes in absorption with the increase of concentration indicate that studied compounds do not aggregate by intermolecular stacking at the experimental conditions used. Emission, absorption maxima, and the corresponding molar extinction coefficients (ε) of all studied compounds are summarized in Table 1. The excitation spectra correspond to compounds’ absorption spectra in the area where emission and excitation spectrum do not overlap.
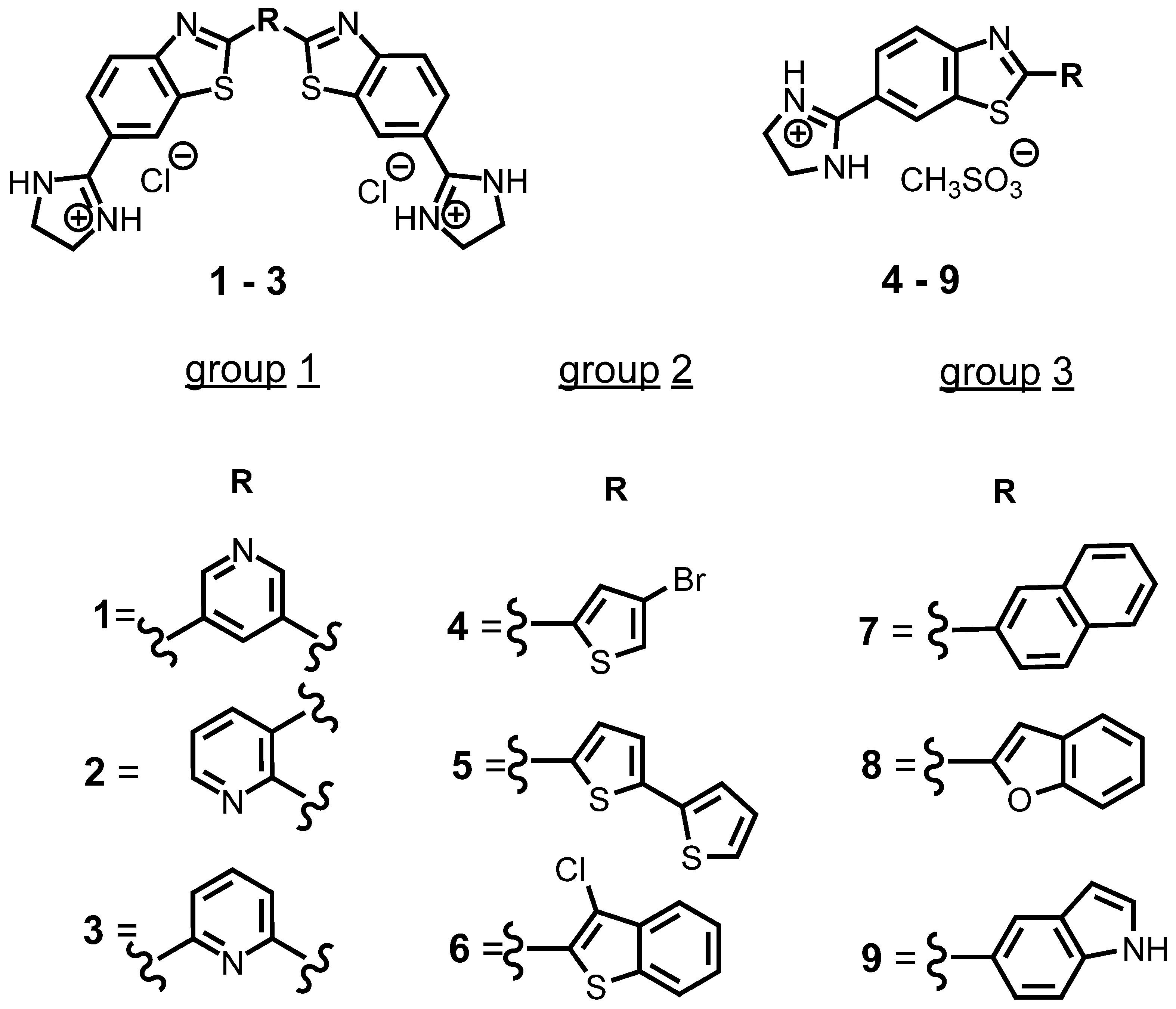
Scheme 1. Structures of benzothiazole derivatives.
Table 1. Electronic absorption data a, absorption maxima, and fluorescence data of benzothiazoles 1–9.
| Compd | UV/Vis λmax (nm) |
ε × 103 /mmol−1 cm2 |
Emission (nm) | Stokes Shift × 10−18 J |
Φf c |
|---|---|---|---|---|---|
| 1 | 330 | 32.1 | 379 | 4.1 | 0.06 |
| 2 | 285/320 b | 31.7/20.6 | 450 | 1.2/1.5 | 0.13 |
| 3 | 330 | 30.7 | 385 | 3.6 | 0.13 |
| 4 | 343 | 44.9 | 416 | 2.7 | 0.37 |
| 5 | 391 | 77.9 | 497 | 1.9 | 0.69 |
| 6 | 342 | 25.1 | 430 | 2.3 | 0.20 |
| 7 | 325 | 35.4 | 444 | 1.7 | 0.38 |
| 8 | 355 | 36.6 | 430 | 2.6 | 0.54 |
| 9 | 343 | 24.2 | 442 | 2.0 | 0.11 |
a Sodium cacodylate buffer, I = 0.05 mol dm−3, pH = 7.0; b in fluorescence titrations λmax was 320 nm; c Absolute fluorescence quantum yield was determined by integrating sphere SC-30, Edinburgh Inst., for argon-purged solutions; QY ± 0.5 °C. [37].
3. Study of Interactions of Benzothiazoles with Nucleic Acids in Aqueous Medium
3.1. Competition Dialysis Assay with 1–9
Competition dialysis assay is a potent and valuable quantitative tool for examining compounds of interest that recognize selective structure/sequence of nucleic acid. In this method, an array of nucleic acid sequences and structures is used. Each is placed in a separate MINI dialysis unit fixed in a flotation dialysis supporter inside a glass container and dialyzed against a ligand solution. The free ligand solution is the same for all the structures, but when equilibrium is reached, each of the structure will bind the ligand according to its binding affinity [29][38].
Nine previously synthesized [34][35][36] benzothiazole compounds with structural changes (Figure 2) in position 2 of the imidazole-based benzothiazole core were used to determine the effect of these modifications on the ability of compounds to interact with different DNA and RNA structures.
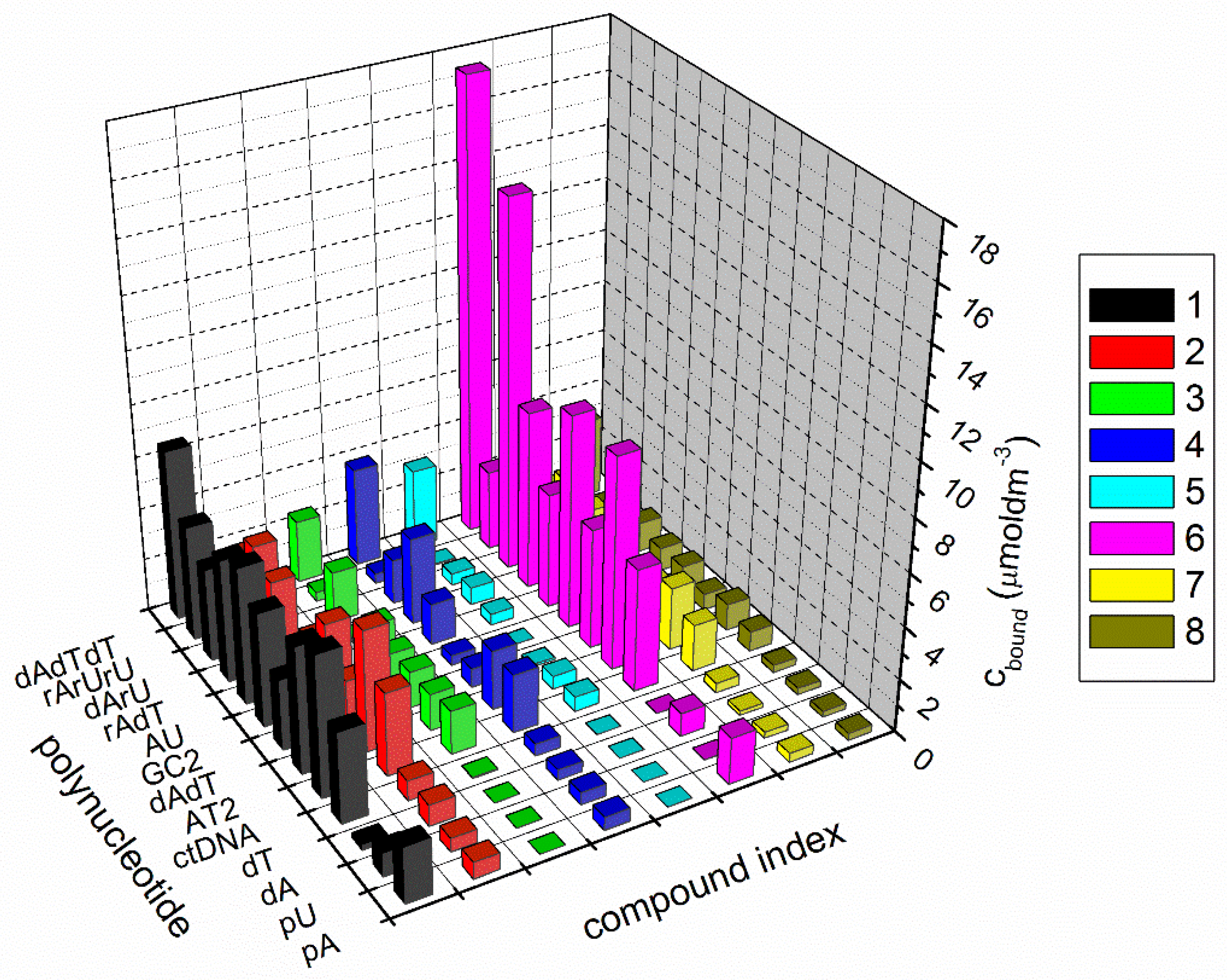
Figure 2. Summary of competition dialysis results with eight compounds binding to 13 nucleic acid structures and sequences (cbound = concentration of ligand bound to each nucleic acid in µM); sodium cacodylate buffer, I = 0.05 mol dm−3, pH = 7, and +1 mM EDTA.
However, the interaction of eight compounds with 13 different nucleic acid structures was studied (Figure 2): single-stranded and double-stranded polynucleotides, DNA:RNA hybrids, and DNA and RNA triplexes. The interactions of compound 9 with polynucleotides were not further characterized, as compound 9 aggregated in the aqueous solution during the experiment.
The interaction of compounds with double-stranded polynucleotides depended on the base composition and secondary structure. Mostly, compounds demonstrated strong binding to DNA triplex ATT. Especially for 5, and even more so, 6, where binding with ATT triplex was stronger than with the double-stranded DNA, RNA, and DNA:RNA hybrids. As the amount of ligand bound to each nucleic acid structure (Cbound) is directly proportional to the ligand binding affinity, it is clear that 1 and 6 displayed the highest affinities toward the majority of the nucleic acid structures. Regarding DNA:RNA hybrids, 6 demonstrated the strongest binding to poly dA−poly rU, while 1 bound slightly better to poly rA−poly dT than to poly dA−poly rU. Only 1 demonstrated preferential binding to single-stranded poly dT, while both 1 and 6 exhibited stronger binding to poly rA.
Two metrics were used, the specificity sum, SS and the ratio Cmax/SS, to gain information about the structural selectivity and compound affinity. To calculate the specificity sum, the binding data first need to be normalized relative to the maximal amount bound (Cmax) to any of the structures in this assay. Then normalized amounts for each nucleic acid structure in the assay were simply summed. As 13 nucleic acid structures were used in this experiment, the SS ratio can range from 1, which denotes the binding to only one nucleic acid structure, to 13, which means an equal binding to all structures. According to Figure 3, the best structural selectivity demonstrated 5 and 6. Values for the specificity sum for all studied compounds are shown in Figure 3.
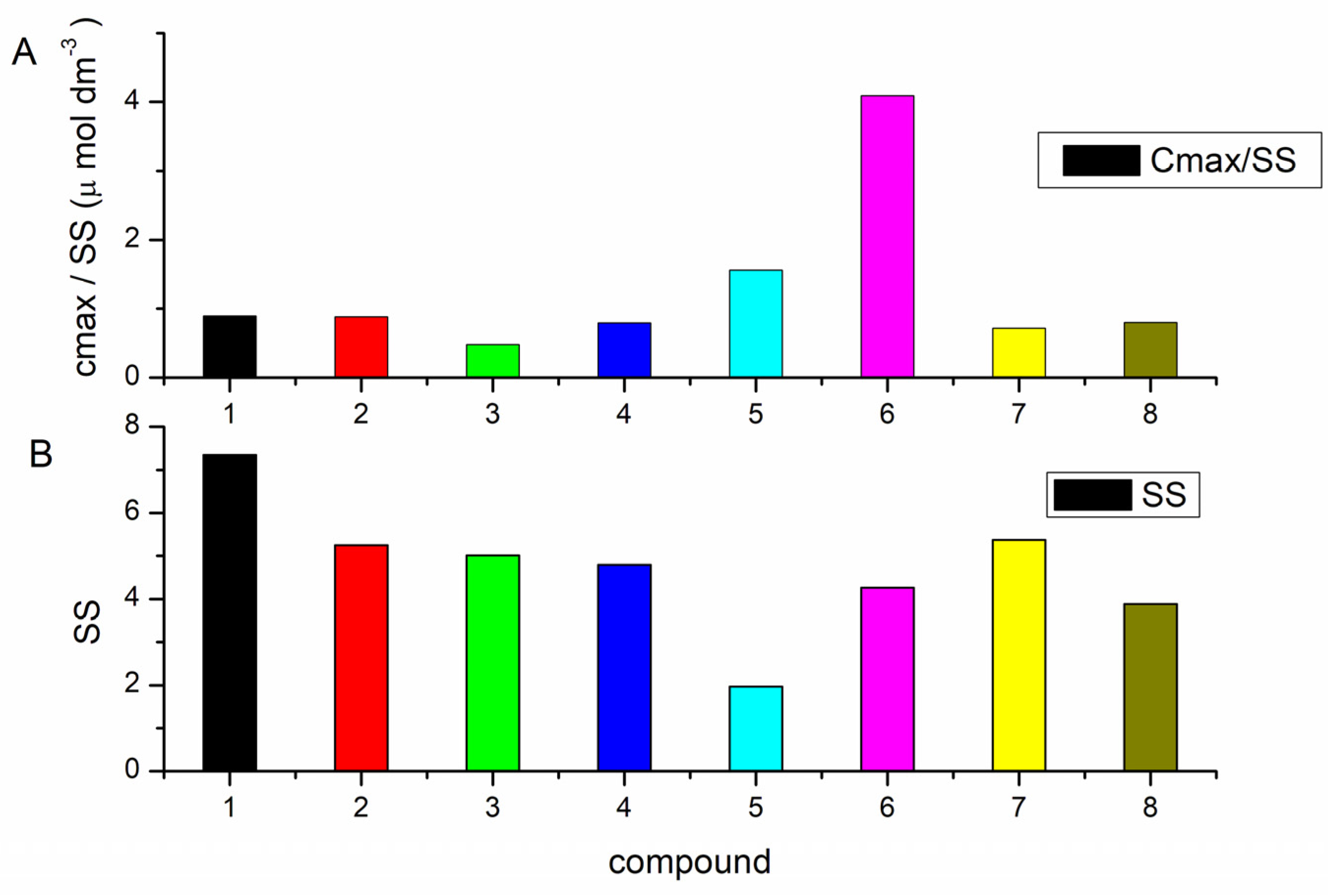
Figure 3. Ratio cmax/SS (A) and specific sum SS (B) for 8 benzothiazole compounds.
Ratio Cmax/SS refers to both affinity and selectivity. This ratio is directly proportional to binding affinity. Thus, if Cmax is large (high binding affinity) and SS is small (high selectivity), a high value of Cmax/SS will be obtained and vice versa [29][38].
Identification of compounds with the best-combined selectivity and affinity can be obtained by comparison of SS and Cmax/SS values. Based on these metrics, 5 and 6 were identified as compounds with the greatest combined selectivity and affinity. Despite the largest SS value, 1 was also selected for further characterization with polynucleotides, as it demonstrated relatively higher Cbound values (amount of ligand bound to each nucleic acid structure), determined for ds- and triplex polynucleotides than other compounds. All three compounds demonstrated selectivity for ATT triplex. Additionally, 6 exhibited the highest affinity toward poly dA−poly rU, while 1 displayed a higher affinity toward poly rA–poly dT.
3.2. Fluorescence Spectroscopy and Isothermal Titration Calorimetry
Isothermal titration calorimetry and fluorescence spectroscopy have been used to characterize the ligand binding to nucleic acid structures [39][40]. According to the results of the competition dialysis assay (affinities and selectivities), several complexes were selected for detailed characterization: 1-ATT (Figure 4), 1-poly rA−poly dT, 6-ATT, 6-poly dA−poly rU, and 5-ATT (Table 2).
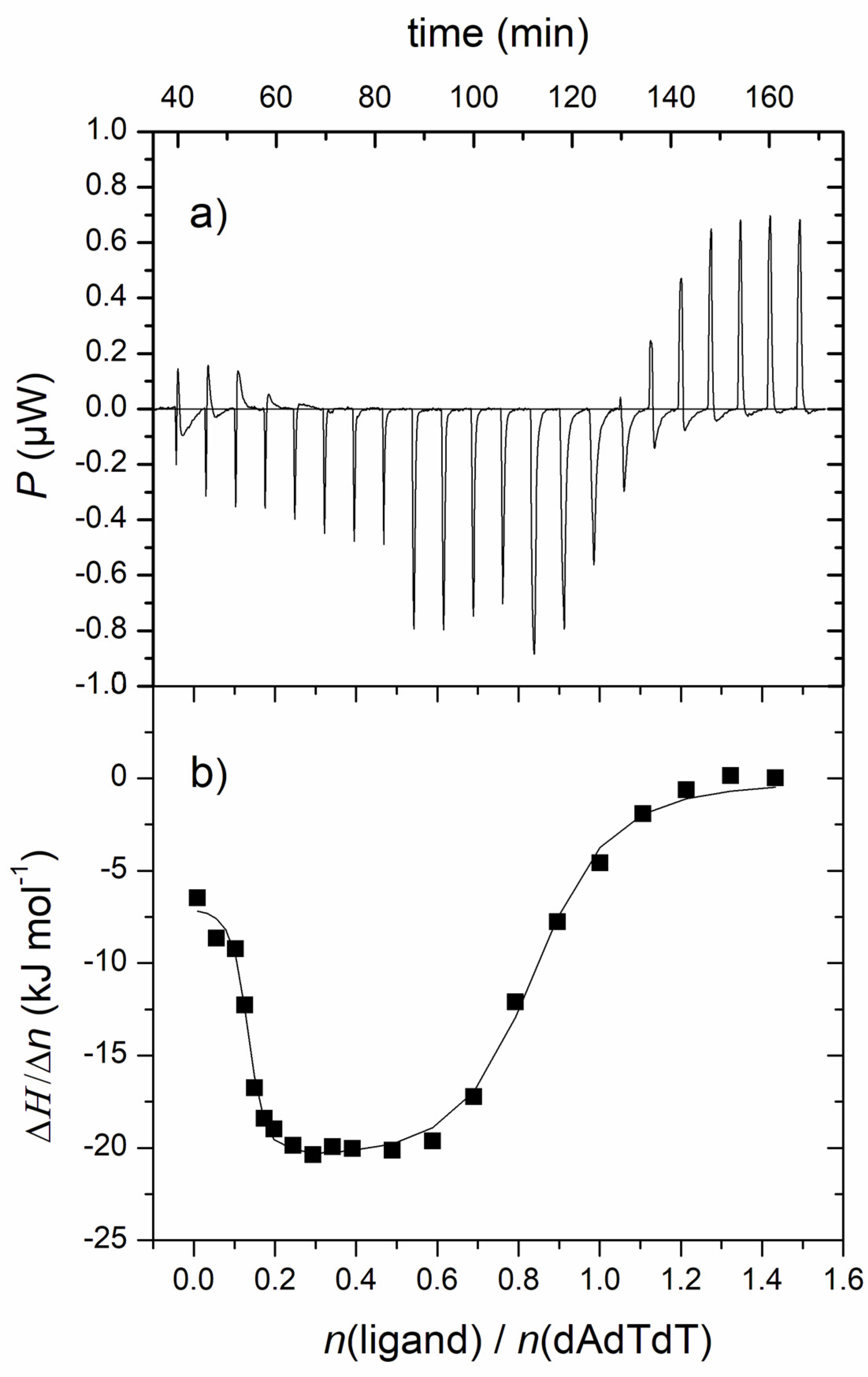
Figure 4. (a) Raw titration data from the single injection of 1 into a solution of ATT triplex; (b) ITC experiment of ATT triplex titrated with 1; experimental data (■) and calculated fit (–) for model of two sets of sites. [ATT] = 3.0 × 10−5 M; pH = 7.0, Na-cacodylate buffer, I = 0.05 mol dm−3 + 1 mM EDTA.
Table 2. Data parameters obtained during nonlinear regression (model: one and two sets of sites) for ITC titration for 1-ATT, 1-poly rA−poly dT complex. Binding constants (logKs) a and ratios n a ([bound compound]/[polynucleotide phosphate]) calculated from the fluorescence titrations for ligand-nucleic acid complex d.
| Complex | n1/n2 | log Ks1/Ks2 | ΔrH1,2°/kJ mol−1 | TΔr1,2S°/kJ mol−1 | ΔrG1,2°/kJ mol−1 |
|---|---|---|---|---|---|
| 1-ATT | 0.7/0.1 | 6.5/8.7 | −21.1/−6.8 | 15.7/43.3 | −36.8/−50.1 |
| n | log Ks | ΔrH°/kJ mol−1 | TΔrS°/kJ mol−1 | ΔrG°/kJ mol−1 | |
| 1-rA-dT | 0.1 | 7.8 | −6.3 | 38.4 | −44.7 |
| 6-ATT b | 0.7 | 7.6 | - c | - c | - c |
| 6-dA-rU b | 0.8 | 8.0 | - c | - c | - c |
a Processing of titration data using Scatchard [41][42] equation gave values of ratio n [bound compound]/[polynucleotide]; correlation coefficients were >0.99 for most of calculated Ks; accuracy of n ± 20%, consequently log Ks values vary in the same order of magnitude; b I/I0 for 6-ATT and 6-poly dA−poly rU = 0.2 and 0.3; I0—starting fluorescence intensity of compound; I—fluorescence intensity of compound/polynucleotide complex calculated by Scatchard [41][42] equation; c Binding affinities were obtained by fluorescence spectroscopy. d All measurements were done at pH = 7.0 (buffer sodium cacodylate, I = 0.05 mol dm−3).
Binding affinities of 5 and 6 could not be calculated by ITC titrations due to the aggregation of these compounds in the ITC method concentration range that interfered with obtaining reliable results. For 6, binding affinities were assessed by fluorescence spectroscopy, which enabled measurements in lower sample concentrations. The fluorescence changes of 5 in titration with ATT triplex were too small for the accurate calculation of the binding constant.
ITC titration experiments resulted in mostly negative peaks, indicating that the binding processes were exothermic (Figure 4). The resulting data for the 1-poly rA−poly dT complex was fitted to a single-site binding model, while data for 1-ATT was fitted to model two sets of sites by using a nonlinear least square method (Table 2). The analysis of ITC experiments of compound 1 with poly rA−poly dT and ATT triplex demonstrated high and similar binding affinities (log Ks, Table 2), which is consistent with the results from competitive dialysis assay.
Compound 1 demonstrated two types of binding in titration with ATT triplex (Figure 4). The first binding event was characterized by a higher binding constant than the second type of binding. The first binding event is an entropically guided process probably accompanied by the release of bound water molecules from the polynucleotide groove to the bulk [39]. The second type of binding, characterized by a higher ratio N, is an enthalpy-driven process accompanied by an increase in the number of hydrogen bonds, aromatic stacking, electrostatic interactions, and van der Waals interactions, followed by a large favorable entropy contribution. The interaction of 1 with poly rA−poly dT was, similar to its complex with ATT, characterized by positive (favorable) binding entropy and weak negative enthalpy (Table 2), indicating an entropically driven process.
The groove binding is usually entropically favorable and slightly endothermic, resulting from the release of relatively highly ordered water molecules surrounding the apolar surfaces to the bulk. The enthalpy contribution to the free energy is associated with the overall increase of bonding (hydrogen bonds, ionic, electrostatic and van der Waals interactions, and polarization of the interacting groups) [43].
An analysis of the fluorescence data of 6 with poly dA−poly rU and ATT polynucleotides gave high binding constants and rather high ratios, N, for both complexes. Unlike 6, 5 induced small emission changes upon binding to ATT triplex, thus disabling calculation of the stability constant.
3.3. Thermal Melting Experiments and RNase H Assay
Thermal denaturation is an additional method for monitoring binding to nucleic acids [44]. Stabilization of nucleic acid structure induced by small molecules causes an increase in the melting temperature of that structure. In a typical experiment, a ligand is monitored against one nucleic acid structure at conditions close to the equimolar ligand/nucleic acid ratio. The melting of mixtures, a relatively straightforward extension of the typical thermal melting experiment design, enables an evaluation of the stabilization effect of the ligand against a number of different nucleic acid structures. If there is an excess of nucleic acid over the compound where the binding sites are not fully saturated, a preference toward sequence or structure can be established [38].
In this experiment, ligand selectivity was studied with a mixture of four different polynucleotides, DNA (poly dA–poly dT), RNA (poly rA–poly rU), and two DNA:RNA hybrids (poly rA–poly dT and poly dA–poly rU) (Figure 5, Table 3).
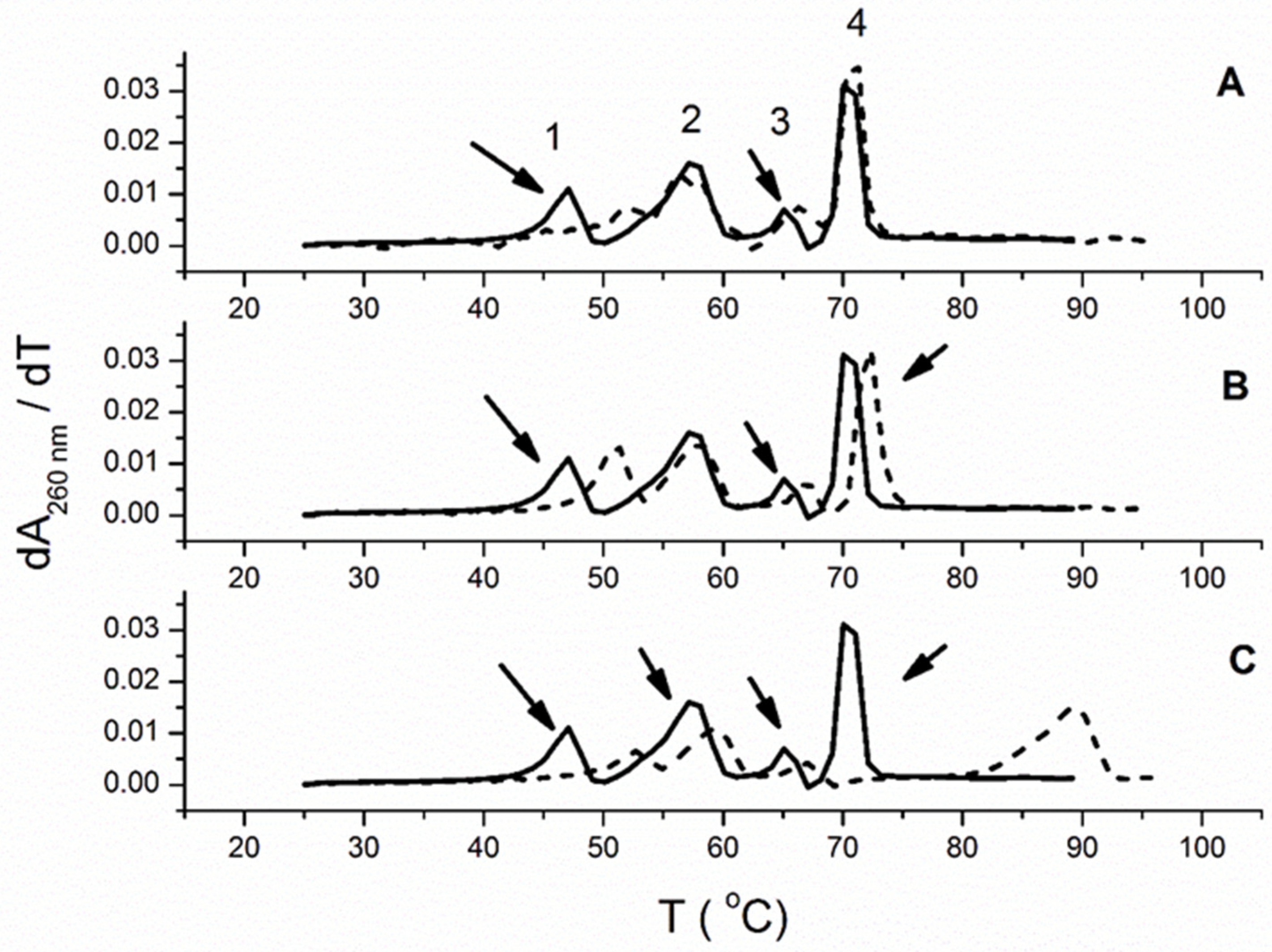
Figure 5. Melting profiles of polynucleotide mixtures (first derivative profile of absorbance versus temperature). Each panel shows the melting of a mixture of 4 polynucleotides: a DNA:RNA hybrid [poly dA−poly rU; peak 1], RNA [poly rA−poly rU; peak 2], an RNA:DNA hybrid [poly rA−poly dT; peak 3] and DNA [poly dA-poly dT; peak 4] as the solid black line (−). The concentration of each polynucleotide structure was 20 μM (bp); total polynucleotide concentration is 80 μM (bp). The dashed line (- - -) in each panel shows the effect of the addition of ligand. (A) 6 (B) 5 (C) 1 all at 2 μM (50 mM sodium cacodylate buffer, 50 mM NaCl, and 1 mM EDTA).
Table 3. The ΔTm a values (°C) of studied mixture of four polynucleotides (poly rA−poly dT, poly rA−poly rU, poly dA−poly rU, and poly dA-poly dT) b upon addition of ratio r = 0.025 c of 1, 5, and 6 d.
| ΔTm a | |||
|---|---|---|---|
| Polynucleotide | 1 | 5 | 6 |
| poly dA−poly rU | 5.6 | 4 | 5.5 |
| poly rA−poly rU | 2.2 | 1 | 0 |
| poly rA−poly dT | 1.7 | 1.7 | 1.5 |
| poly dA-poly dT | 19.1 | 2.1 | 0 |
a ΔTm = Tm (complex polynucleotide-dye) − Tm (polynucleotide); error in ΔTm: ±0.5 °C; b Tm values for mixture of polynucleotides without compound, poly dA−poly rU = 47 °C; poly rA−poly rU = 57.4 °C; poly rA−poly dT = 65.3 °C; poly dA-poly dT = 70.6 °C.; c r = [compound]/[polynucleotide]. d All measurements were done at pH 7.0 (sodium cacodylate buffer with NaCl, I = 0.1 mol dm−3 + 1 mM EDTA).
The compounds 1 and 5 demonstrated the stabilization effect of all studied ds-polynucleotides (Table 3, Figure 5). In addition, these two compounds demonstrated a better stabilization effect of poly dA−poly rU than of poly rA−poly dT. Interestingly, among ds-nucleic structures, 6 almost exclusively stabilized poly dA−poly rU. It has been demonstrated that the stability of hybrid duplexes, composed of homopurine and homopyrimidine strands, depends on several factors, such as the percentage of deoxyribo(pyrimidine), i.e., d(Py) content in each of the strands, the oligomeric length and the percentage of (A)n:(T or U)n content [45]. Thus, for example, the hybrid duplex containing the DNA purine strand and the RNA pyrimidine strand d(Pu):r(Py), such as poly dA−poly rU, demonstrates much less thermal stability compared to the r(Pu):d(Py) composition, such as poly rA−poly dT. This could be a reason for the higher stabilization effect of dArU, in comparison to rAdT, induced by 1, 5, and 6. Among ds-polynucleotides, 1 demonstrated the most significant stabilization effect with poly dA−poly rU and especially poly dA−poly dT (Table 3). It should be emphasized that 1 possesses a higher number of net positive charges than 5 and 6 (1, +2; 5, +1; 6, +1). However, only 6 demonstrated selective stabilization of the hybrid duplex, poly dA−poly rU, in a mixture of double-stranded polynucleotides.
As all three ligands stabilized poly rA−poly dT and poly dA−poly rU, researchers performed a spectrophotometric RNase H assay to evaluate the capability of 1, 5, and 6 to inhibit RNase H [29]. The cleaving of the RNA strand from a DNA:RNA hybrid duplex was followed by an increase in A260 (Figure 6).
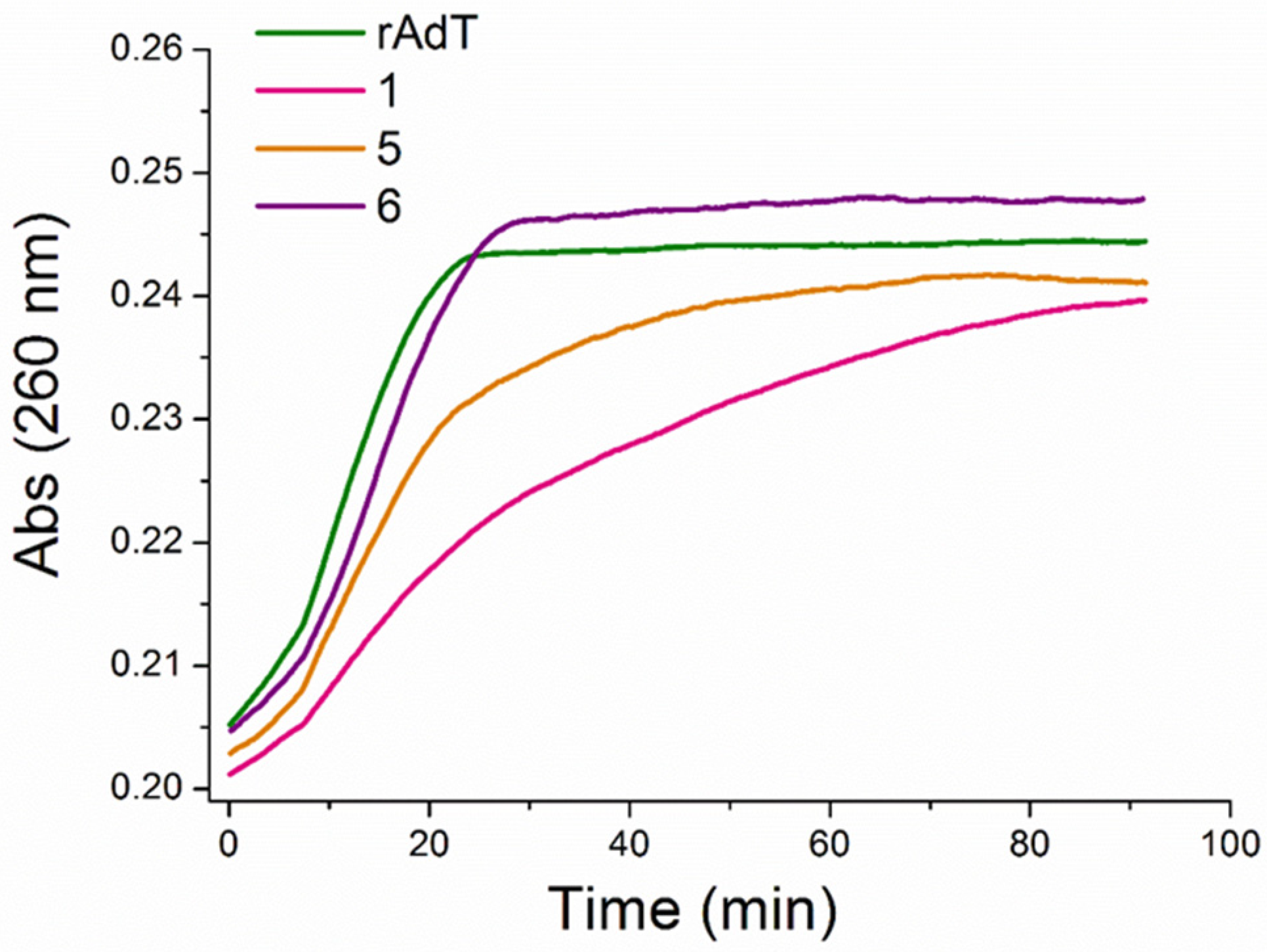
Figure 6. Inhibition of RNase H digestion of poly rA−poly dT by 1, 5, and 6; concentration of nucleic acid hybrid structure was 10 μM (bp); 1, 5, and 6, all at 3 μM.
The best inhibition activity of the RNase H digestion of poly rA−poly dT demonstrated benzothiazole 1. The data from a thermal melting of polynucleotide mixtures show a good correlation with the results of the RNase H assay. The RNase H assay was utilized here as an additional method to the thermal melting of polynucleotide mixtures. It provides information on which ligands can be biologically relevant as RNase H inhibitors and deserves detailed investigation regarding that.
The melting profile of free ATT in sodium cacodylate buffer (50 mM sodium cacodylate buffer, 50 mM NaCl, and 1 mM EDTA) demonstrated biphasic transition (Figure 7, Table 4).
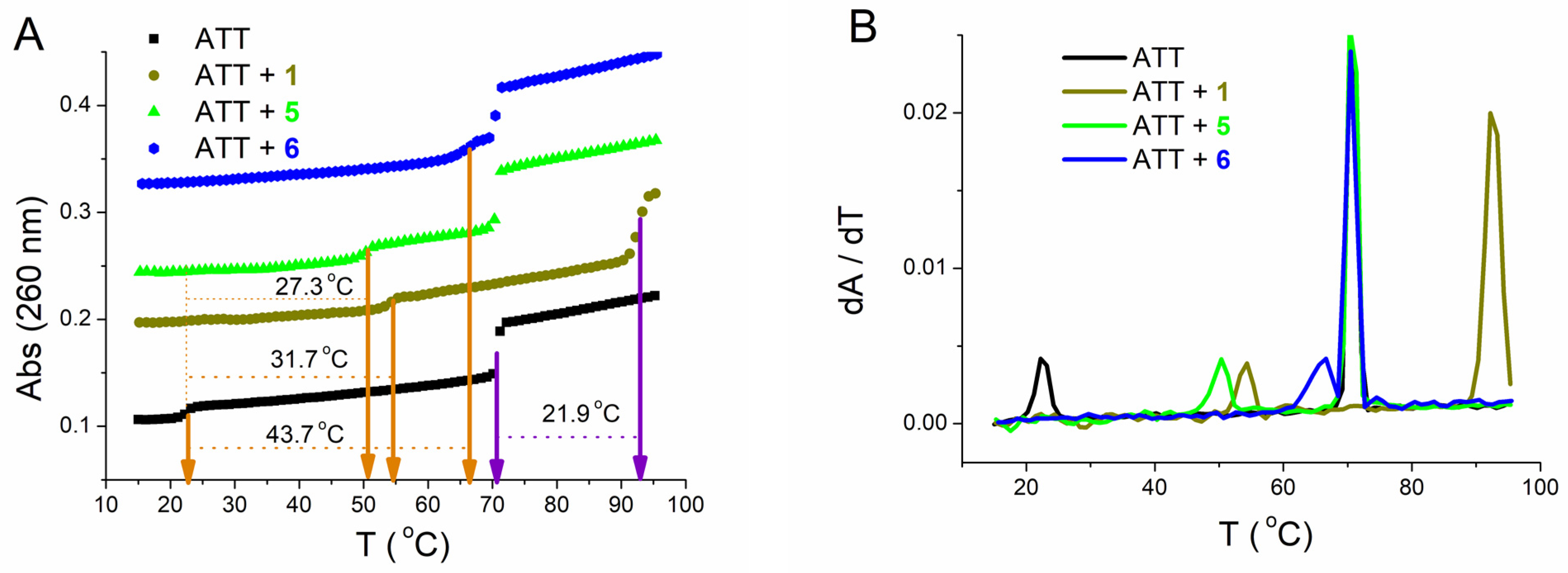
Figure 7. (A): Melting curve of ATT triplex upon addition of ratio, r ([compound/[polynucleotide]) = 0.1 of 1, 5, and 6 at pH = 7.0 (sodium cacodylate buffer with NaCl, I = 0.1 mol dm−3 + 1 mM EDTA; orange arrows—first transition, violet arrows—second transition); (B): First derivative of absorbance at 260 nm in dependence of temperature.
Table 4. The ΔTm a values (°C) of ATT triplex b upon addition of ratio c r = [compound]/[polynucleotide] of 1, 5, and 6 d.
| ΔTm a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 | 6 | |||||||
| cr | 0.1 | 0.2 | 0.3 | 0.1 | 0.2 | 0.3 | 0.025 | 0.05 | 0.1 |
| 31.7/21.9 | 32.5/23.9 | - | 27.3/0 | 30.1/0 | 32.8/0 | 36.7/0 | 42.0/0 | 43.7/0 | |
a ΔTm = Tm (complex polynucleotide-dye) − Tm (polynucleotide); error in ΔTm: ±0.5 °C; b Tm values for biphasic thermal denaturation profile of ATT without compound, Tm1 = 22.6; Tm2 = 70.7; c r = [compound]/[polynucleotide]; d All measurements were done at pH 7.0 (sodium cacodylate buffer with NaCl, I = 0.1 mol dm−3 + 1 mM EDTA).
The first transition was at Tm1 = 22.6 °C and corresponds to dissociation of the third strand (poly dT with Hoogsteen base pairs from the major groove). The second transition was at Tm2 = 70.7 °C and corresponds to dissociation of Watson–Crick base pairs of double-stranded poly dA-poly dT [46][47]. Albeit 1 stabilized both the ATT triplex and poly dA−poly dT duplex, the higher stabilization effect was demonstrated with the ATT triplex. Unlike 1, 5 and 6 increased Tm1, while Tm2 did not change. Particularly interesting was compound 6, which stabilized ATT significantly even at very low ratios, r (Figure 7). Data from this experiment for poly dA−poly dT (Table 4) duplex agrees very well with those for the same polynucleotide obtained in the melting of mixtures experiment (Table 3).
3.4. Circular Dichroism (CD) Experiments
Based on competition dialysis results, researchers further proceeded with the characterization of selected complexes by CD spectroscopy. Electronic circular dichroism (ECD) is highly sensitive toward conformational changes in the helical structure of DNA and RNA and their complexes with small molecules [48][49]. In addition, an induced CD spectrum (ICD) that can arise from the interaction of achiral small molecules such as 1, 5, and 6 with nucleic acids, could be very informative of the binding modes. Researchers monitored changes in CD spectra upon the interaction of 1, 5, and 6 with representatives of the B-helix family, ctDNA with a mixed base pair composition, and with poly dA−poly dT characterized by a much narrower and deeper minor groove in comparison to other common B-helices. As a model of A-helical structure, researchers used poly rA−poly rU (ds-RNA) characterized by a wide and shallow minor groove and deep and narrow major groove [50][51]. Interactions were also studied with two double-stranded DNA:RNA hybrids, poly dA−poly rU, and poly rA−poly dT and the DNA triple helix, ATT. While NMR, Raman, and X-ray fiber diffraction studies suggest B-like conformation for poly rA-poly dT in high humidity conditions, it seems that the global conformation of poly dA-poly rU is considerably more affected by the ribopyrimidine strand, resulting in an A-type helix closer in conformation to the A-form of RNA [52][53]. Further, NMR and IR data suggest that structural characteristics of triplexes resemble B-DNA much more than A-DNA [54][55][56].
The addition of 1, 5, and 6 to polynucleotides mainly caused a decrease of CD intensity of DNA and RNA polynucleotides at their maximal values (ctDNA at 275 nm, AT-DNA, AU-RNA, and DNA:RNA hybrids at 260 nm) (Figure 8); however, induced CD spectra of these compounds, which changed with increasing ratio, r, were more informational regarding their modes of binding.
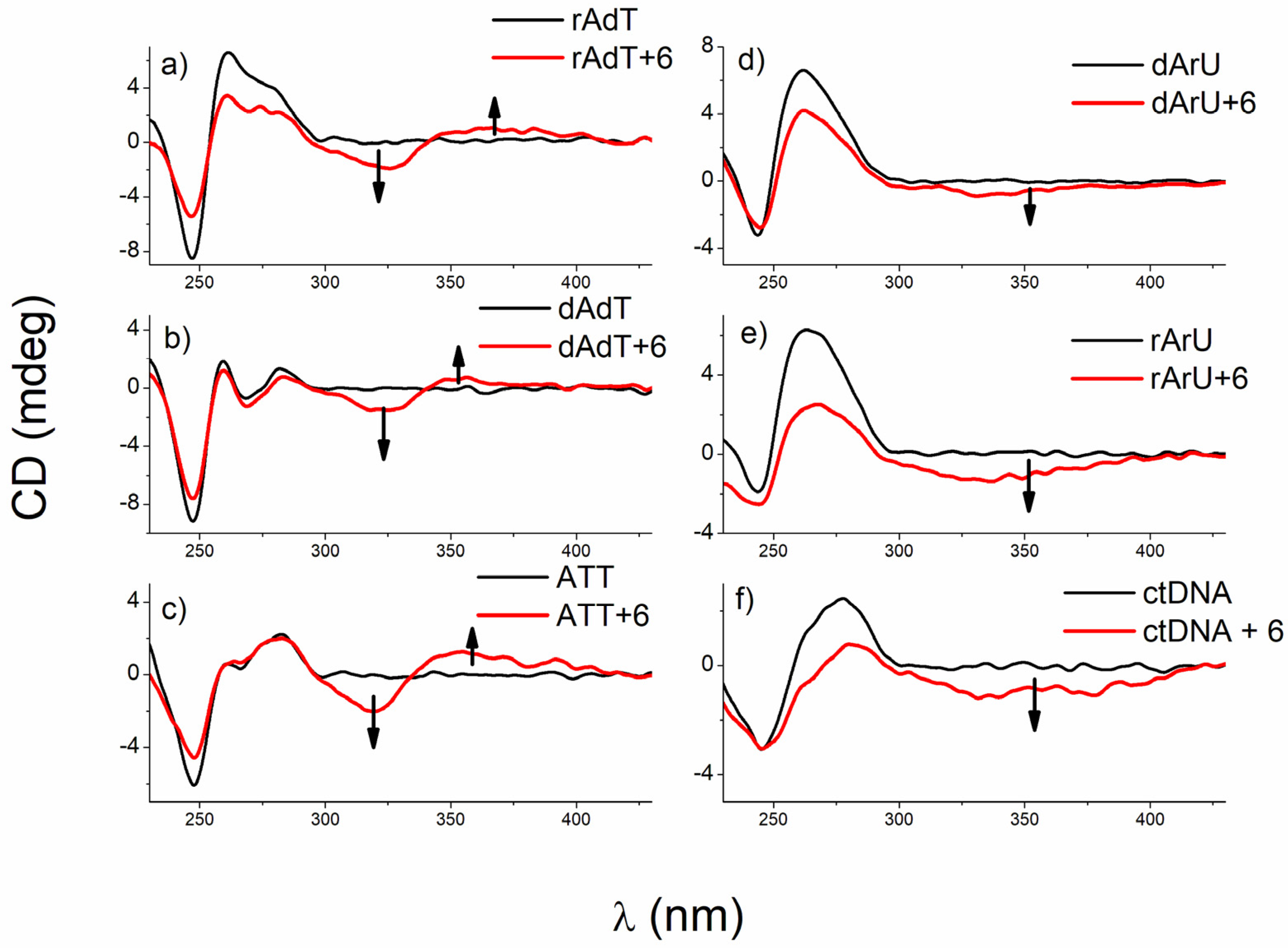
Figure 8. CD titrations of (a) poly rA−poly dT, (b) poly dA−poly dT, (c) ATT triplex, (d) poly dA−poly rU, (e) poly rA−poly rU, and (f) ctDNA (c = 3.0 × 10−5 mol dm−3) with 6 at molar ratios r = [compound]/[polynucleotide] = 0.3 (pH = 7.0, buffer sodium cacodylate, I = 0.05 mol dm−3 + 1 mM EDTA for all titrations except poly dA−poly rU (I = 0.2 mol dm−3 + 1 mM EDTA)).
At r ≤ 0.1, 6 caused either negligible or weak negative ICD signals (around 320 nm) with all polynucleotides. However, at r > 0.1, this compound differentiates between B- and A-type helices by the mode of binding (Figure 8). Negative ICD signals support intercalation to A-type helices, poly dA−poly rU and poly rA−poly rU, whereas an appearance of a bisignate CD signal implied the formation of 6 dimers most probably inside the minor groove of poly rA−poly dT, poly dA−poly dT, and ATT triplex. Interactions of 6 were additionally investigated with ATT 26mer by fluorimetric and CD spectroscopy to estimate the influence of the ATT polymer length on the binding strength. Processing of titration data gave ratio n and binding constant (log Ks = 7.4, n = 0.7) comparable to that obtained with the ATT polynucleotide (Table 2). Regarding CD titration of 6 with ATT 26mer, a similar bisignate ICD signal, as in the CD titration with ATT polynucleotide, was noticed, suggesting that ATT polymer length did not influence the mode of binding.
Unexpectedly, 6 did not bind in the form of dimers to ctDNA (B-helix), instead, changes (negative ICD signals, Figure 8) were indicative for intercalation, which can probably be related to the composition of ctDNA containing 42% of GC basepairs beside AT base pairs.
Unlike 6, 5 did not differentiate among polynucleotide conformations. Its addition caused a rise of bisignate ICD signals with all studied nucleic acid structures. Such an effect supports a formation of dimers (minor groove of ATT triplex and major groove of poly rA-poly rU) or larger aggregates, similar to those observed with poly dA-poly dT and ctDNA.
On the other hand, bis-benzothiazolyl-pyridine 1, which is sterically more demanding compared to 5 and 6, provoked a strong positive ICD band (at 340 nm) at r ≤ 0.1, implying binding within the minor groove of ctDNA, poly dA−poly dT, and ATT triplex (Figure 8). At ratios higher than r = 0.1, an excess of 6 molecules cannot accommodate inside the minor groove so well; instead, 6 forms aggregate along the polynucleotide backbones. Negative ICD signals (around 340 nm) at r ≤ 0.1 point towards intercalative binding to poly rA−poly rU and both hybrids. An increase in ICD intensity with an increase of r (r > 0.1) suggests aggregation of 6 along the polynucleotide surfaces. Further, a clear isodichroic point (λ = 253 nm) observed for 1 and poly rA−poly dT strongly suggests one dominant interaction mode of this compound with the DNA chiral axis.
Due to the strong interaction of 1 with the ATT triplex, researchers decided to examine interactions of this triplex with its regioisomer 3 to see whether the position of benzothiazole-imidazolinyl chains on pyridine ring affected them. Interestingly, compound 3, unlike its regioisomer 1, induced a strong increase in the intensity of CD spectra of ATT triplex, poly dA−poly dT, and ctDNA (ctDNA at 275 nm, AT-DNA at 260 and 282 nm, and ATT at 282 nm) compared to the intrinsic CD bands of these polynucleotides, and in addition, strong positive ICD bands located around 350 nm (Figure 9).
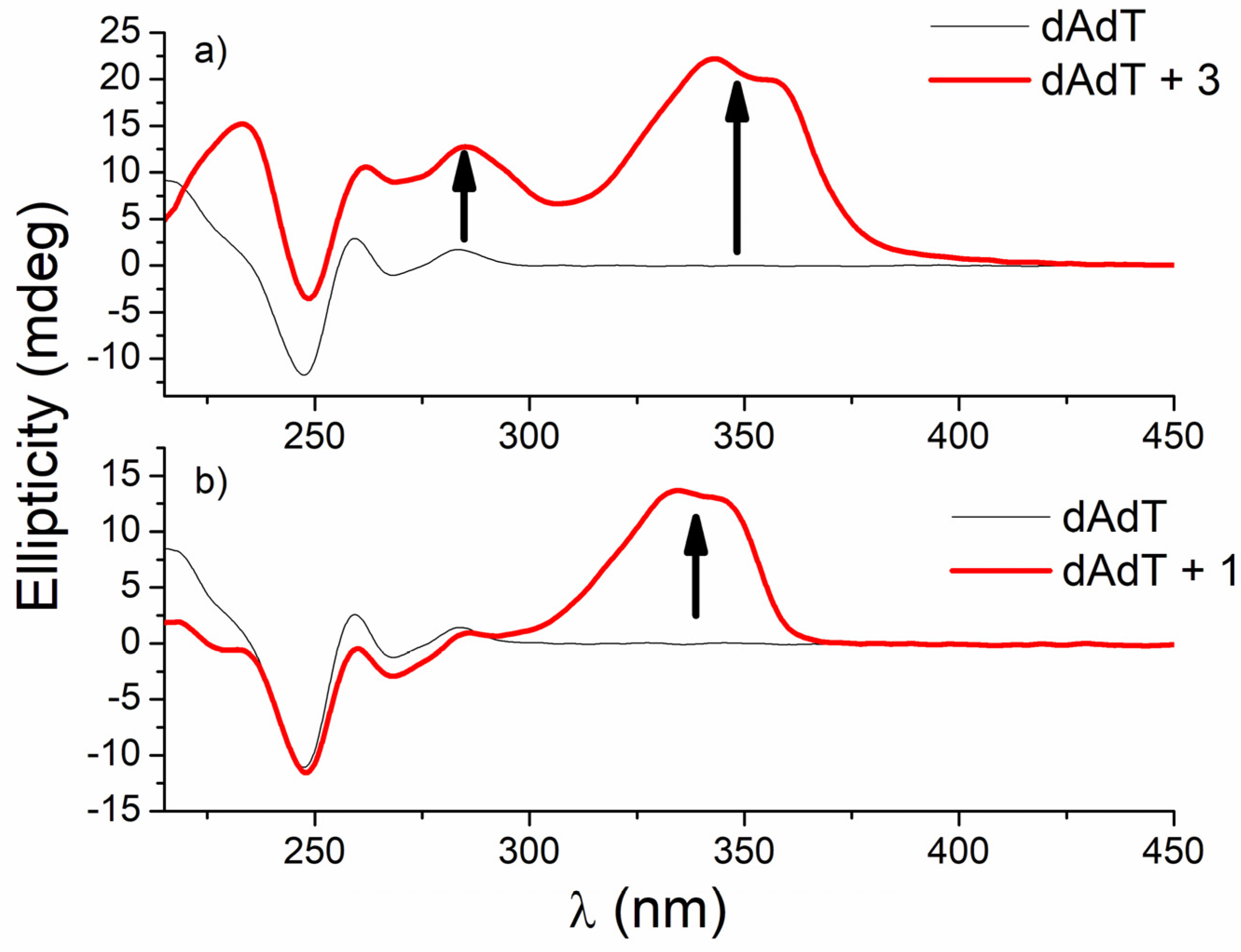
Figure 9. CD titrations of AT-DNA duplex (dAdT) (c = 3.0 × 10−5 mol dm−3) with 3 (a) and 1 (b) at molar ratios r = [compound]/[polynucleotide] = 0.4 and 0.1, respectively (pH = 7.0, buffer sodium cacodylate, I = 0.05 mol dm−3 + 1 mM EDTA).
Similar strong changes in CD spectra of polynucleotides were noticed with bis-polyaza pyridinophane derivatives that, similarly to compound 3, consist of two chains attached to pyridine as a central unit [57]. The reason that compound 3 induced condensation-like changes, while compound 1 did not, is probably the position of chains on the pyridine ring. Unlike 1, 3 and bis-polyaza pyridinophane derivatives have two chains attached at the same positions on the pyridine ring (2 and 6 positions).
3.5. Molecular Modeling
The binding of 6 dimers inside the minor groove of the ATT triplex was also examined by molecular modeling (Figure 10). The mode of binding of 6 with the ATT triple structure suggested by the spectroscopic methods was consistent with the results obtained by molecular modeling.
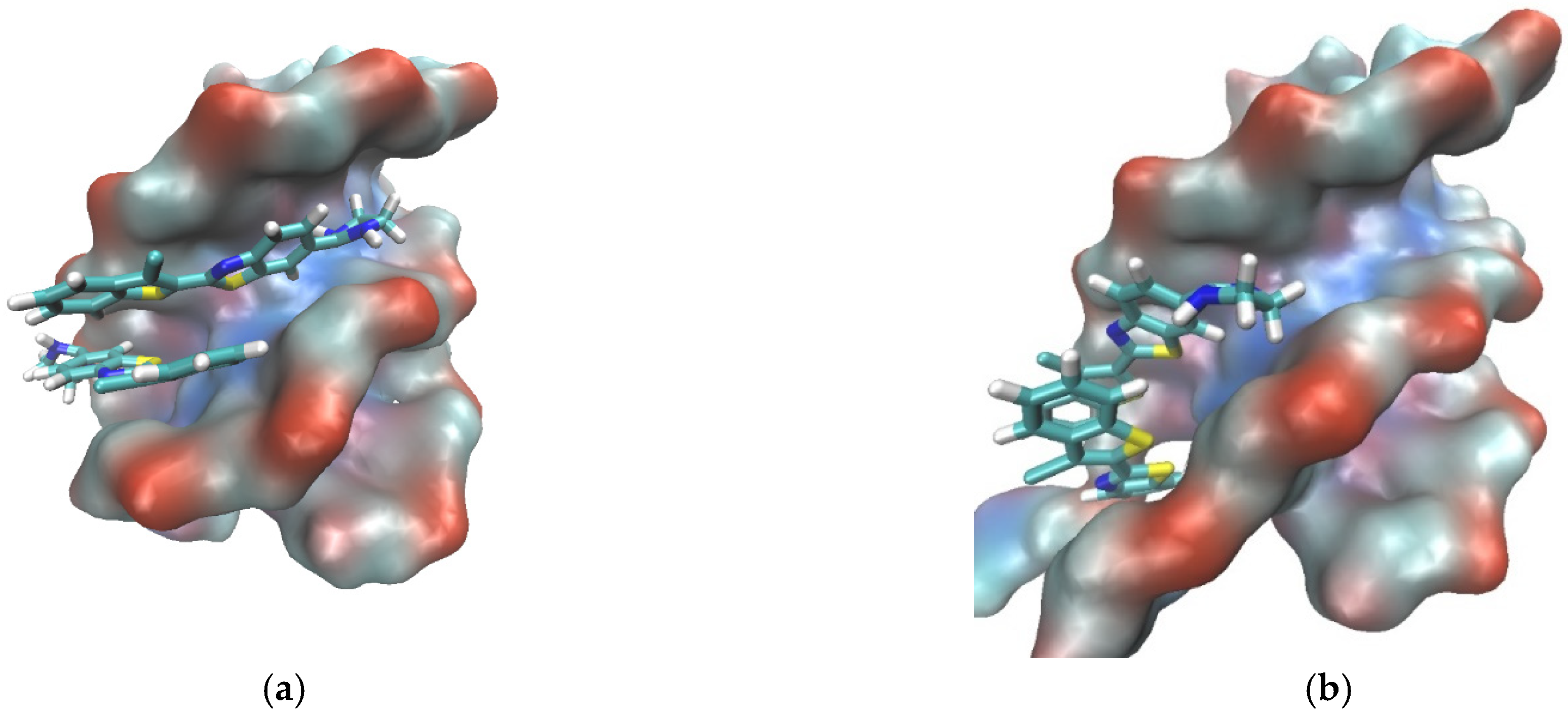
Figure 10. A complex between ATT (DNA triplex) and dimer of 6 obtained after 200 ns of MD simulation in water. ATT is represented by its solvent accessible surface and ligand is given in stick representation. (complexes were built in PyMOL, wherein the initial position of ligands was determined from spectroscopic data. Parametrization was performed by ANTECHAMBER [58] and Leap, the modules available within AMBER16 suite of programs [59][60] using GAFF [61] for the ligands and (a) bsc1 [62] and (b) OL15 [60] for ATT. Neutralized and solvated complexes were minimized, equilibrated, and simulated for 200 ns using the programs sander and pmemd.
References
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Cancer 2002, 2, 188–200.
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Demeunynck, M., Bailly, C., Wilson, W.D., Eds.; Wiley-VCH: Weinheim, Germany, 2003.
- Escudé, C.; Nguyen, C.H.; Kukreti, S.; Janin, Y.; Sun, J.-S.; Bisagni, E.; Garestier, T.; Hélène, C. Rational design of a triple helix-specific intercalating ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 3591–3596.
- Arya, D.P. New Approaches Toward Recognition of Nucleic Acid Triple Helices. Acc. Chem. Res. 2011, 44, 134–146.
- Nguyen, T.Q.N.; Lim, K.W.; Phan, A.T. A Dual-Specific Targeting Approach Based on the Simultaneous Recognition of Duplex and Quadruplex Motifs. Sci. Rep. 2017, 7, 11969.
- Le, D.D.; Di Antonio, M.; Chan, L.K.M.; Balasubramanian, S. G-quadruplex ligands exhibit differential G-tetrad selectivity. Chem. Commun. 2015, 51, 8048–8050.
- Ranjan, N.; Davis, E.; Xue, L.; Arya, D.P. Dual recognition of the human telomeric G-quadruplex by a neomycin–anthraquinone conjugate. Chem. Commun. 2013, 49, 5796–5798.
- Van Dyke, M.W.; Ohyama, T. Do DNA Triple Helices or Quadruplexes Have a Role in Transcription? In DNA Conformation and Transcription; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2005; pp. 105–126.
- Nadal, A.; Eritja, R.; Esteve, T.; Pla, M. “Parallel” and “antiparallel tail-clamps” increase the efficiency of triplex formation with structured DNA and RNA targets. ChemBioChem 2005, 6, 1034–1042.
- Wang, G.; Seidman, M.M.; Glazer, P.M. Mutagenesis in Mammalian Cells Induced by Triple Helix Formation and Transcription-Coupled Repair. Science 1996, 271, 802–805.
- Jain, A.; Wang, G.; Vasquez, K.M. DNA triple helices: Biological consequences and therapeutic potential. Biochimie 2008, 90, 1117–1130.
- Xi, H.; Kumar, S.; Dosen-Micovic, L.; Arya, D.P. Calorimetric and spectroscopic studies of aminoglycoside binding to AT-rich DNA triple helices. Biochimie 2010, 92, 514–529.
- Sharma, S.K.; Frase, W. Selectivity of a bromoacridine containing fuorophore for triplex DNA. Mon. Chem. 2021, 152, 1013–1016.
- Xue, L.; Xi, H.; Kumar, S.; Gray, D.; Davis, E.; Hamilton, P.; Skriba, M.; Arya, D.P. Probing the Recognition Surface of a DNA Triplex: Binding Studies with Intercalator−Neomycin Conjugates. Biochemistry 2010, 49, 5540–5552.
- Mergny, J.L.; Duval-Valentin, G.; Nguyen, C.H.; Perrouault, L.; Faucon, B.; Rougée, M.; Montenay-Garestier, T.; Bisagni, E.; Hélène, C. Triple Helix-Specific Ligands. Science 1992, 256, 1681–1684.
- Houlard, M.; Artus, J.; Leguillier, T.; Vandormael-Pournin, S.; Cohen-Tannoudji, M. DNA-RNA hybrids contribute to the replication dependent genomic instability induced byOmcg1deficiency. Cell Cycle 2011, 10, 108–117.
- Balk, B.; Dees, M.; Bender, K.; Luke, B. The differential processing of telomeres in response to increased telomeric transcription and RNA–DNA hybrid accumulation. RNA Biol. 2014, 11, 95–100.
- Stuckey, R.; García-Rodríguez, N.; Aguilera, A.; Wellinger, R.E. Role for RNA:DNA hybrids in origin-independent replication priming in a eukaryotic system. Proc. Natl. Acad. Sci. USA 2015, 112, 5779–5784.
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205.
- Li, T.-K.; Barbieri, C.M.; Lin, H.-C.; Rabson, A.B.; Yang, G.; Fan, Y.; Gaffney, B.L.; Jones, R.A.; Pilch, D.S. Drug Targeting of HIV-1 RNA·DNA Hybrid Structures: Thermodynamics of Recognition and Impact on Reverse Transcriptase-Mediated Ribonuclease H Activity and Viral Replication. Biochemistry 2004, 43, 9732–9742.
- Lombraña, R.; Almeida, R.; Alvarez, A.; Gómez, M. R-loops and initiation of DNA replication in human cells: A missing link? Front. Genet. 2015, 6, 158.
- Saretzki, G. Telomerase inhibition as cancer therapy. Cancer Lett. 2003, 194, 209–219.
- Mergny, J.-L.; Lacroix, L.; Teulade-Fichou, M.-P.; Hounsou, C.; Guittat, L.; Hoarau, M.; Arimondo, P.B.; Vigneron, J.-P.; Lehn, J.-M.; Riou, J.-F.; et al. Telomerase inhibitors based on quadruplex ligands selected by a fluorescence assay. Proc. Natl. Acad. Sci. USA 2001, 98, 3062–3067.
- Su, H.-P.; Yan, Y.; Prasad, G.S.; Smith, R.F.; Daniels, C.L.; Abeywickrema, P.D.; Reid, J.C.; Loughran, H.M.; Kornienko, M.; Sharma, S.; et al. Structural Basis for the Inhibition of RNase H Activity of HIV-1 Reverse Transcriptase by RNase H Active Site-Directed Inhibitors. J. Virol. 2010, 84, 7625–7633.
- Tramontano, E.; Di Santo, R. HIV-1 RT-associated RNase H function inhibitors: Recent advances in drug development. Curr. Med. Chem. 2010, 17, 2837–2853.
- Ren, J.; Qu, X.; Dattagupta, N.; Chaires, J. Molecular Recognition of a RNA:DNA Hybrid Structure. J. Am. Chem. Soc. 2001, 123, 6742–6743.
- Shaw, N.N.; Arya, D.P. Recognition of the unique structure of DNA:RNA hybrids. Biochimie 2008, 90, 1026–1039.
- West, C.; Francis, R.; Friedman, S.H. Small molecule/Nucleic acid affinity chromatography: Application for the identification of telomerase inhibitors which target its key RNA/DNA heteroduplex. Bioorganic Med. Chem. Lett. 2001, 11, 2727–2730.
- Wheelhouse, R.T.; Chaires, J.B. Drug binding to DNA x RNA hybrid structures. Methods Mol. Biol. 2010, 613, 55–70.
- Shi, X.; Chaires, J.B. Sequence- and structural-selective nucleic acid binding revealed by the melting of mixtures. Nucleic Acids Res. 2006, 34, e14.
- Shaw, N.N.; Xi, H.; Arya, D.P. Molecular recognition of a DNA:RNA hybrid: Sub-nanomolar binding by a neomycin–methidium conjugate. Bioorganic Med. Chem. Lett. 2008, 18, 4142–4145.
- Xi, H.; Davis, E.; Ranjan, N.; Xue, L.; Hyde-Volpe, D.; Arya, D.P. Thermodynamics of Nucleic Acid “Shape Readout” by an Aminosugar. Biochemistry 2011, 50, 9088–9113.
- Wheelhouse, R.T.; Garbett, N.C.; Buurma, N.J.; Chaires, J. Probing the Molecular Recognition of a DNA⋅RNA Hybrid Duplex. Angew. Chem. Int. Ed. 2010, 49, 3207–3210.
- Racané, L.; Kraljević Pavelić, S.; Ratkaj, I.; Stepanić, V.; Pavelić, K.; Tralić-Kulenović, V.; Karminski-Zamola, G. Synthesis and antiproliferative evaluation of some new amidino-substituted bis-benzothiazolyl-pyridines and pyrazine. Eur. J. Med. Chem. 2012, 55, 108–116.
- Racané, L.; Sedić, M.; Ilić, N.; Aleksić, M.; Pavelić, S.K.; Karminski-Zamola, G. Novel 2-Thienyl- and 2-Benzothienyl-Substituted 6-(2-Imidazolinyl)Benzothiazoles: Synthesis; in vitro Evaluation of Antitumor Effects and Assessment of Mitochondrial Toxicity. Anti-Cancer Agents Med. Chem. 2017, 17, 57–66.
- Racané, L.; Ptiček, L.; Sedić, M.; Grbčić, P.; Pavelić, S.K.; Bertosa, B.; Sović, I.; Karminski-Zamola, G. Eco-friendly synthesis, in vitro anti-proliferative evaluation, and 3D-QSAR analysis of a novel series of monocationic 2-aryl/heteroaryl-substituted 6-(2-imidazolinyl)benzothiazole mesylates. Mol. Divers. 2018, 22, 723–741.
- Košćak, M.; Krošl, I.; Žinić, B.; Piantanida, I. Fluorescent Analogues of FRH Peptide: Cu(II) Binding and Interactions with ds-DNA/RNA. Chemosensors 2022, 10, 34.
- Ragazzon, P.; Chaires, J.B. Use of competition dialysis in the discovery of G-quadruplex selective ligands. Methods 2007, 43, 313–323.
- Bronowska, A.K. Thermodynamics of Ligand-Protein Interactions: Implications for Molecular Design. In Thermodynamics—Interaction Studies—Solids, Liquids and Gases; IntechOpen: London, UK, 2011.
- Perozzo, R.; Folkers, G.; Scapozza, L. Thermodynamics of Protein–Ligand Interactions: History, Presence, and Future Aspects. J. Recept. Signal Transduct. 2004, 24, 1–52.
- Scatchard, G. The Attractions of Proteins for Small Molecules and Ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672.
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489.
- Chaires, J.B. A thermodynamic signature for drug–DNA binding mode. Arch. Biochem. Biophys. 2006, 453, 26–31.
- Mergny, J.-L.; Lacroix, L. Analysis of Thermal Melting Curves. Oligonucleotides 2003, 13, 515–537.
- Lesnik, E.A.; Freier, S.M. Relative Thermodynamic Stability of DNA, RNA, and DNA:RNA Hybrid Duplexes: Relationship with Base Composition and Structure. Biochemistry 1995, 34, 10807–10815.
- Choi, B.-H.; Yeo, G.; Jung, J.; Lee, B.W.; Han, S.; Cho, T. A Thermodynamic Investigation into the Stabilization of Poly(dA)· 2 Triple Helical DNA by Various Divalent Metal Ions. Bull. Korean Chem. Soc. 2009, 30, 2691–2696.
- Arya, D.P.; Coffee, R.L.; Willis, B.; Abramovitch, A.I. Aminoglycoside−Nucleic Acid Interactions: Remarkable Stabilization of DNA and RNA Triple Helices by Neomycin. J. Am. Chem. Soc. 2001, 123, 5385–5395.
- Berova, N.; Nakanishi, K.; Woody, R. Circular Dichroism Principles and Applications, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000.
- Eriksson, M.; Nordén, B. Linear and circular dichroism of drug-nucleic acid complexes. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 2001; Volume 340, pp. 68–98.
- Egli, M.; Saenger, W. Principles of Nucleic Acid Structure; Springer: Berlin/Heidelberg, Germany, 1983.
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; W.H. Freeman: San Francisco, CA, USA, 1980.
- Neidle, S. Oxford Handbook of Nucleic Acid Structure; Oxford University Press: Oxford, UK, 1999.
- Fedoroff, O.Y.; Salazar, M.; Reid, B.R. Structure of a DNA: RNA Hybrid Duplex: Why RNase H Does Not Cleave Pure RNA. J. Mol. Biol. 1993, 233, 509–523.
- Radhakrishnan, I.; Patel, D.J. Solution structure of a pyrimidine·purine·pyrimidine DNA triplex containing T·AT, C+ GC and G·TA triples. Structure 1994, 2, 17–32.
- Raghunathan, G.; Miles, H.T.; Sasisekharan, V. Symmetry and molecular structure of a DNA triple helix: D(T)n.cntdot.d(A)n.cntdot.d(T)n. Biochemistry 1993, 32, 455–462.
- Esguerra, M.; Nilsson, L.; Villa, A. Triple helical DNA in a duplex context and base pair opening. Nucleic Acids Res. 2014, 42, 11329–11338.
- Stojković, M.R.; González-García, J.; Šupljika, F.; Galiana-Roselló, C.; Guijarro, L.; Gazzè, S.A.; Francis, L.W.; Piantanida, I.; García-España, E. Specific and highly efficient condensation of GC and IC DNA by polyaza pyridinophane derivatives. Int. J. Biol. Macromol. 2017, 109, 143–151.
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260.
- Case, D.; Betz, R.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.; Giese, T.J.; Gohlke, H.; Götz, A.; Homeyer, N.; et al. Amber 16; University of California: San Francisco, CA, USA, 2016.
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, I.T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174.
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined ForceField for DNA Simulations. Nat. Methods 2015, 13, 55–58.
More
Information
Subjects:
Biochemical Research Methods
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
4 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No