 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laurence Dubrez | + 3229 word(s) | 3229 | 2022-02-21 07:20:36 | | | |
| 2 | Beatrix Zheng | Meta information modification | 3229 | 2022-03-10 13:08:58 | | | | |
| 3 | Beatrix Zheng | + 222 word(s) | 3451 | 2022-03-14 07:11:06 | | | | |
| 4 | Beatrix Zheng | Meta information modification | 3451 | 2022-03-18 02:40:13 | | |
Video Upload Options
Cellular inhibitor of apoptosis 1 (cIAP1) is a cell signaling regulator of the IAP family. Through its E3-ubiquitine ligase activity, it has the ability to activate intracellular signaling pathways, modify signal transduction pathways by changing protein-protein interaction networks, and stop signal transduction by promoting the degradation of critical components of signaling pathways. Thus, cIAP1 appears to be a potent determinant of the response of cells, enabling their rapid adaptation to changing environmental conditions or intra- or extracellular stresses. It is expressed in almost all tissues, found in the cytoplasm, membrane and/or nucleus of cells. cIAP1 regulates innate immunity by controlling signaling pathways mediated by tumor necrosis factor receptor superfamily (TNFRs), some cytokine receptors and pattern recognition-receptors (PRRs). Although less documented, cIAP1 has also been involved in the regulation of cell migration and in the control of transcriptional programs.
1. Introduction
2. Cytoplasmic Functions of cIAP1
2.1. Role for cIAP1 in Regulating Innate Immunity
2.1.1. Regulation of TNFα Signaling Pathways in Immune and Non-Immune Cells
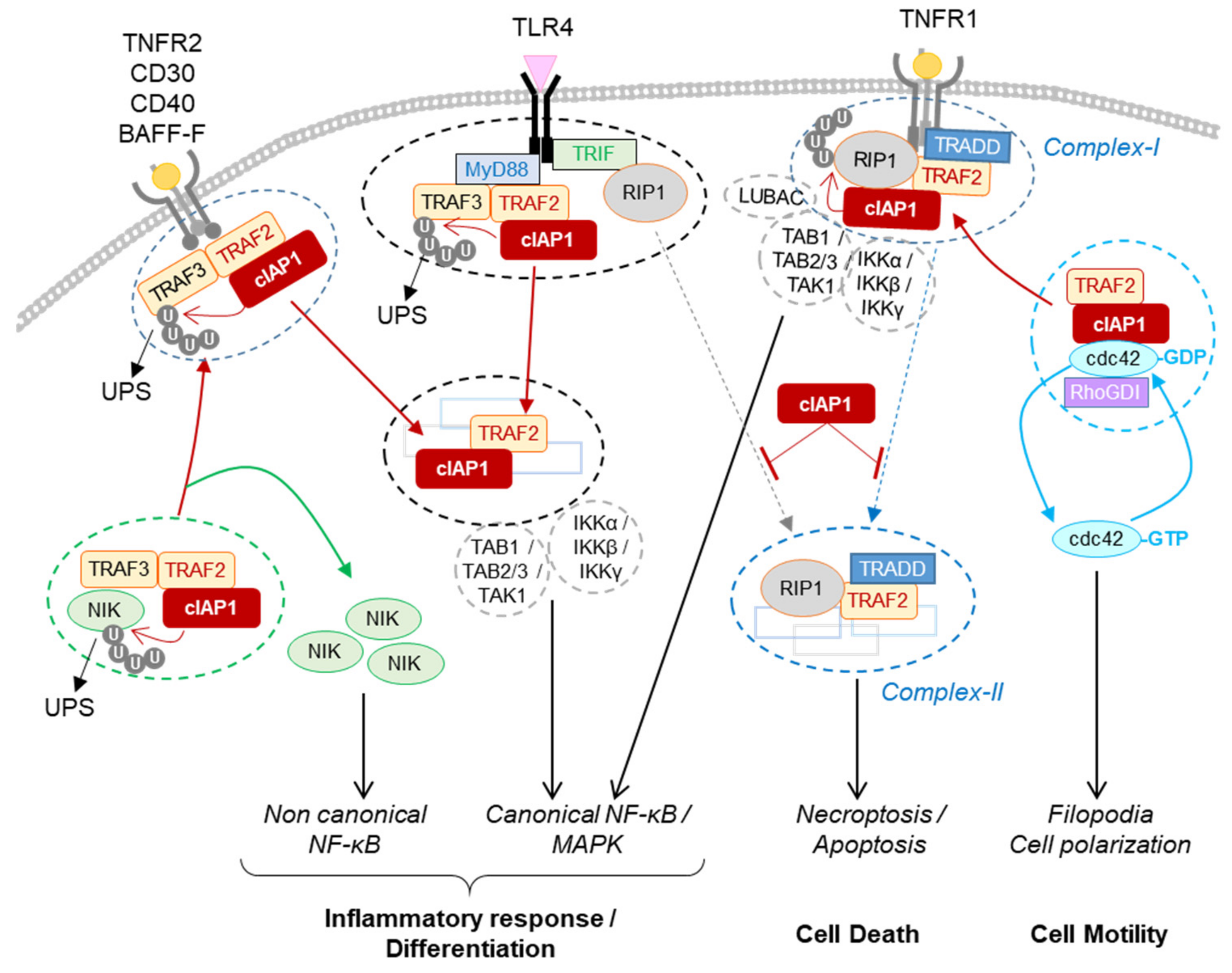
2.1.2. Regulation of the Non-Canonical NF-κB Signaling Pathway in Immune Cells, Osteoclasts and Endothelial Cells
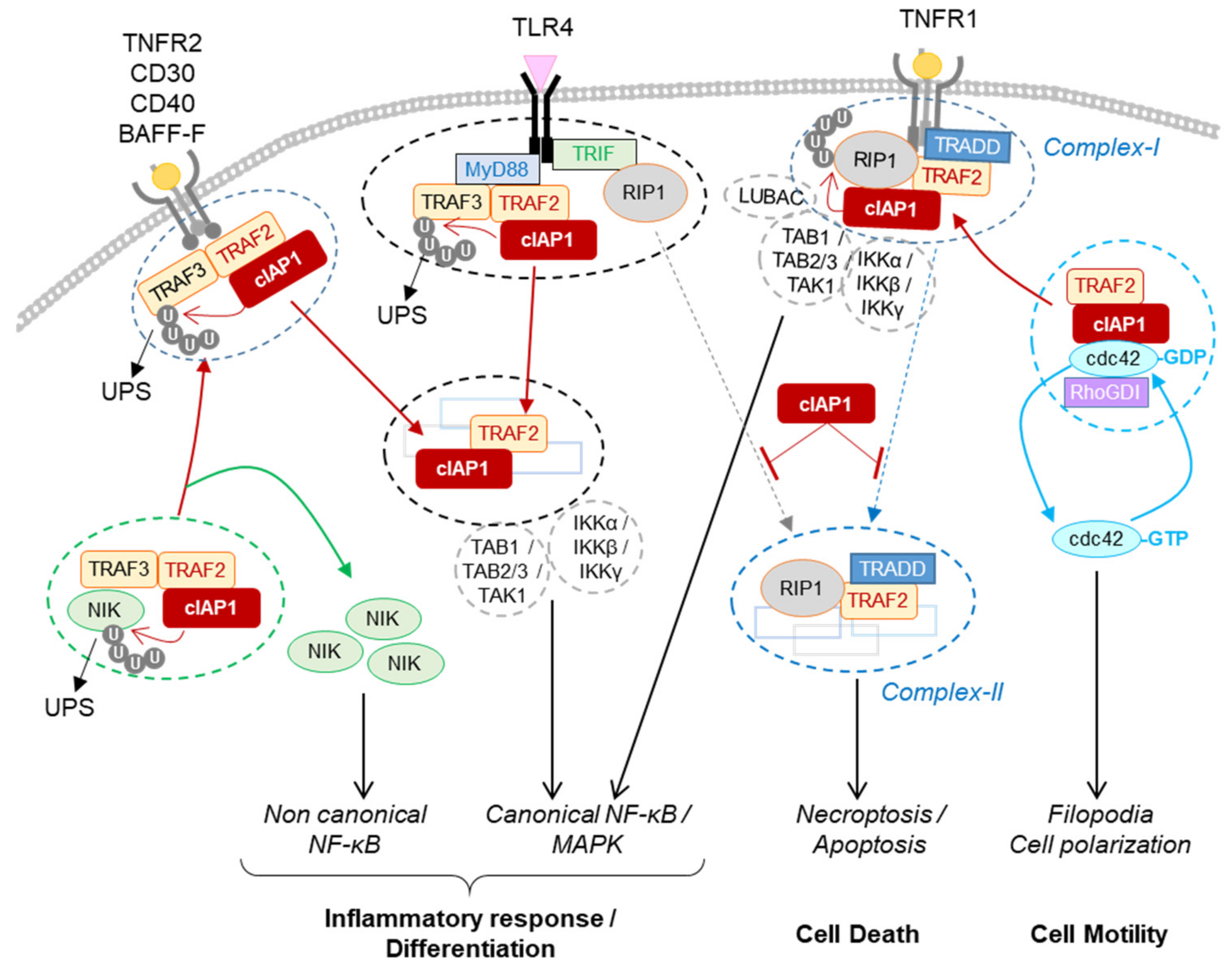
2.1.3. Regulation of PRR Signaling Pathways
2.2. Role for cIAP1 in Cell Motility and Migration
3. Conclusions
References
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174.
- Liston, P.; Roy, N.; Tamai, K.; Lefebvre, C.; Baird, S.; Cherton-Horvat, G.; Farahani, R.; McLean, M.; Ikeda, J.E.; MacKenzie, A.; et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996, 379, 349–353.
- Vucic, D.; Stennicke, H.R.; Pisabarro, M.T.; Salvesen, G.S.; Dixit, V.M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 2000, 10, 1359–1366.
- Vucic, D.; Kaiser, W.J.; Harvey, A.J.; Miller, L.K. Inhibition of reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs). Proc. Natl. Acad. Sci. USA 1997, 94, 10183–10188.
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304.
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998, 273, 7787–7790.
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Datta, P.; Alnemri, E.S.; Shi, Y. Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780.
- Chai, J.; Du, C.; Wu, J.W.; Kyin, S.; Wang, X.; Shi, Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000, 406, 855–862.
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438.
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 2002, 277, 445–454.
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406.
- Uren, A.G.; Pakusch, M.; Hawkins, C.J.; Puls, K.L.; Vaux, D.L. Cloning and expression of apoptosis inhibitory protein homologs that function to inhibit apoptosis and/or bind tumor necrosis factor receptor-associated factors. Proc. Natl. Acad. Sci. USA 1996, 93, 4974–4978.
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461.
- Estornes, Y.; Bertrand, M.J. IAPs, regulators of innate immunity and inflammation. Semin. Cell Dev. Biol. 2015, 39, 106–114.
- Dumetier, B.; Zadoroznyj, A.; Dubrez, L. IAP-Mediated Protein Ubiquitination in Regulating Cell Signaling. Cells 2020, 9, 1118.
- Wang, D.; Berglund, A.E.; Kenchappa, R.S.; MacAulay, R.J.; Mulé, J.J.; Etame, A.B. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Sci. Rep. 2017, 7, 9350.
- Allègre, J.; Cartier, J.; Glorian, V.; Droin, N.; Dumetier, B.; Kayaci, C.; Berthelet, J.; Gemble, S.; Vuillier, C.; Maillet, L.; et al. E2F1 binds to the peptide-binding groove within the BIR3 domain of cIAP1 and requires cIAP1 for chromatin binding. PLoS ONE 2018, 13, e0206253.
- Warnakulasuriyarachchi, D.; Cerquozzi, S.; Cheung, H.H.; Holcik, M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J. Biol. Chem. 2004, 279, 17148–17157.
- Riley, A.; Jordan, L.E.; Holcik, M. Distinct 5′ UTRs regulate XIAP expression under normal growth conditions and during cellular stress. Nucleic Acids Res. 2010, 38, 4665–4674.
- Van Eden, M.E.; Byrd, M.P.; Sherrill, K.W.; Lloyd, R.E. Translation of cellular inhibitor of apoptosis protein 1 (c-IAP1) mRNA is IRES mediated and regulated during cell stress. RNA 2004, 10, 469–481.
- Didelot, C.; Lanneau, D.; Brunet, M.; Bouchot, A.; Cartier, J.; Jacquel, A.; Ducoroy, P.; Cathelin, S.; Decologne, N.; Chiosis, G.; et al. Interaction of heat-shock protein 90 beta isoform (HSP90 beta) with cellular inhibitor of apoptosis 1 (c-IAP1) is required for cell differentiation. Cell Death Differ. 2008, 15, 859–866.
- Marivin, A.; Berthelet, J.; Plenchette, S.; Dubrez, L. The Inhibitor of Apoptosis (IAPs) in Adaptive Response to Cellular Stress. Cells 2012, 1, 711–737.
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267.
- Ma, O.; Cai, W.W.; Zender, L.; Dayaram, T.; Shen, J.; Herron, A.J.; Lowe, S.W.; Man, T.K.; Lau, C.C.; Donehower, L.A. MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res. 2009, 69, 2559–2567.
- Cheng, L.; Zhou, Z.; Flesken-Nikitin, A.; Toshkov, I.A.; Wang, W.; Camps, J.; Ried, T.; Nikitin, A.Y. Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene 2010, 29, 5700–5711.
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Res 2019, 8.
- Tseng, W.Y.; Huang, Y.S.; Lin, H.H.; Luo, S.F.; McCann, F.; McNamee, K.; Clanchy, F.; Williams, R. TNFR signalling and its clinical implications. Cytokine 2018, 101, 19–25.
- Zhang, M.; Wang, J.; Jia, L.; Huang, J.; He, C.; Hu, F.; Yuan, L.; Wang, G.; Yu, M.; Li, Z. Transmembrane TNF-α promotes activation-induced cell death by forward and reverse signaling. Oncotarget 2017, 8, 63799–63812.
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922.
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65.
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461.
- Witt, A.; Vucic, D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017, 24, 1160–1171.
- Schwarzer, R.; Laurien, L.; Pasparakis, M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr. Opin. Cell Biol. 2020, 63, 186–193.
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114.
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282.
- Chen, K.W.; Lawlor, K.E.; von Pein, J.B.; Boucher, D.; Gerlic, M.; Croker, B.A.; Bezbradica, J.S.; Vince, J.E.; Schroder, K. Cutting Edge: Blockade of Inhibitor of Apoptosis Proteins Sensitizes Neutrophils to TNF- but Not Lipopolysaccharide-Mediated Cell Death and IL-1β Secretion. J. Immunol. 2018, 200, 3341–3346.
- Grabinger, T.; Bode, K.J.; Demgenski, J.; Seitz, C.; Delgado, M.E.; Kostadinova, F.; Reinhold, C.; Etemadi, N.; Wilhelm, S.; Schweinlin, M.; et al. Inhibitor of Apoptosis Protein-1 Regulates Tumor Necrosis Factor-Mediated Destruction of Intestinal Epithelial Cells. Gastroenterology 2017, 152, 867–879.
- Wong, W.W.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O’Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 2014, 123, 2562–2572.
- Liu, H.; Liao, R.; He, K.; Zhu, X.; Li, P.; Gong, J. The SMAC mimetic birinapant attenuates lipopolysaccharide-induced liver injury by inhibiting the tumor necrosis factor receptor-associated factor 3 degradation in Kupffer cells. Immunol. Lett. 2017, 185, 79–83.
- Gentle, I.E.; Moelter, I.; Lechler, N.; Bambach, S.; Vucikuja, S.; Häcker, G.; Aichele, P. Inhibitors of apoptosis proteins (IAPs) are required for effective T-cell expansion/survival during antiviral immunity in mice. Blood 2014, 123, 659–668.
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 2008, 283, 24295–24299.
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691.
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463.
- Moen, I.N.; Westhrin, M.; Håland, E.; Haug, M.; Nonstad, U.; Klaharn, M.; Standal, T.; Starheim, K.K. Smac-mimetics reduce numbers and viability of human osteoclasts. Cell Death Discov. 2021, 7, 36.
- Zhang, J.; Webster, J.D.; Dugger, D.L.; Goncharov, T.; Roose-Girma, M.; Hung, J.; Kwon, Y.C.; Vucic, D.; Newton, K.; Dixit, V.M. Ubiquitin Ligases cIAP1 and cIAP2 Limit Cell Death to Prevent Inflammation. Cell Rep. 2019, 27, 2679–2689.e2673.
- Anderton, H.; Rickard, J.A.; Varigos, G.A.; Lalaoui, N.; Silke, J. Inhibitor of Apoptosis Proteins (IAPs) Limit RIPK1-Mediated Skin Inflammation. J. Investig. Dermatol. 2017, 137, 2371–2379.
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Graß, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682.
- Annibaldi, A.; Wicky John, S.; Vanden Berghe, T.; Swatek, K.N.; Ruan, J.; Liccardi, G.; Bianchi, K.; Elliott, P.R.; Choi, S.M.; Van Coillie, S.; et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol. Cell 2018, 69, 566–580.e565.
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257.
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr. Biol. 2007, 17, 418–424.
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209.
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227.
- Tang, E.D.; Wang, C.Y.; Xiong, Y.; Guan, K.L. A role for NF-kappaB essential modifier/IkappaB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IkappaB kinase complex by tumor necrosis factor-alpha. J. Biol. Chem. 2003, 278, 37297–37305.
- Borghi, A.; Haegman, M.; Fischer, R.; Carpentier, I.; Bertrand, M.J.M.; Libert, C.; Afonina, I.S.; Beyaert, R. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-κB signaling. Biochem. Pharm. 2018, 153, 292–298.
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370.
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681.
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378.
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85.
- Lee, S.; Challa-Malladi, M.; Bratton, S.B.; Wright, C.W. Nuclear factor-κB-inducing kinase (NIK) contains an amino-terminal inhibitor of apoptosis (IAP)-binding motif (IBM) that potentiates NIK degradation by cellular IAP1 (c-IAP1). J. Biol. Chem. 2014, 289, 30680–30689.
- Csomos, R.A.; Wright, C.W.; Galban, S.; Oetjen, K.A.; Duckett, C.S. Two distinct signalling cascades target the NF-kappaB regulatory factor c-IAP1 for degradation. Biochem. J. 2009, 420, 83–91.
- Matsuzawa, A.; Tseng, P.H.; Vallabhapurapu, S.; Luo, J.L.; Zhang, W.; Wang, H.; Vignali, D.A.; Gallagher, E.; Karin, M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 2008, 321, 663–668.
- Gardam, S.; Turner, V.M.; Anderton, H.; Limaye, S.; Basten, A.; Koentgen, F.; Vaux, D.L.; Silke, J.; Brink, R. Deletion of cIAP1 and cIAP2 in murine B lymphocytes constitutively activates cell survival pathways and inactivates the germinal center response. Blood 2011, 117, 4041–4051.
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Komuves, L.G.; Deshayes, K.; Vucic, D. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci. Signal. 2012, 5, ra22.
- Vince, J.E.; Chau, D.; Callus, B.; Wong, W.W.; Hawkins, C.J.; Schneider, P.; McKinlay, M.; Benetatos, C.A.; Condon, S.M.; Chunduru, S.K.; et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell Biol. 2008, 182, 171–184.
- Hatem, J.; Schrank-Hacker, A.M.; Watt, C.D.; Morrissette, J.J.; Rubin, A.I.; Kim, E.J.; Nasta, S.D.; Wasik, M.A.; Bogusz, A.M. Marginal zone lymphoma-derived interfollicular diffuse large B-cell lymphoma harboring 20q12 chromosomal deletion and missense mutation of BIRC3 gene: A case report. Diagn. Pathol. 2016, 11, 137.
- Rosebeck, S.; Lim, M.S.; Elenitoba-Johnson, K.S.; McAllister-Lucas, L.M.; Lucas, P.C. API2-MALT1 oncoprotein promotes lymphomagenesis via unique program of substrate ubiquitination and proteolysis. World J. Biol. Chem. 2016, 7, 128–137.
- Matthews, G.M.; de Matos Simoes, R.; Dhimolea, E.; Sheffer, M.; Gandolfi, S.; Dashevsky, O.; Sorrell, J.D.; Mitsiades, C.S. NF-κB dysregulation in multiple myeloma. Semin. Cancer Biol. 2016, 39, 68–76.
- Dupoux, A.; Cartier, J.; Cathelin, S.; Filomenko, R.; Solary, E.; Dubrez-Daloz, L. cIAP1-dependent TRAF2 degradation regulates the differentiation of monocytes into macrophages and their response to CD40 ligand. Blood 2009, 113, 175–185.
- Yang, C.; Davis, J.L.; Zeng, R.; Vora, P.; Su, X.; Collins, L.I.; Vangveravong, S.; Mach, R.H.; Piwnica-Worms, D.; Weilbaecher, K.N.; et al. Antagonism of inhibitor of apoptosis proteins increases bone metastasis via unexpected osteoclast activation. Cancer Discov. 2013, 3, 212–223.
- Tseng, P.H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2010, 11, 70–75.
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602.
- Busca, A.; Konarski, Y.; Gajanayaka, N.; O’Hara, S.; Angel, J.; Kozlowski, M.; Kumar, A. cIAP1/2-TRAF2-SHP-1-Src-MyD88 Complex Regulates Lipopolysaccharide-Induced IL-27 Production through NF-κB Activation in Human Macrophages. J. Immunol. 2018, 200, 1593–1606.
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.C. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6, 5930.
- Mathew, S.J.; Haubert, D.; Kronke, M.; Leptin, M. Looking beyond death: A morphogenetic role for the TNF signalling pathway. J. Cell Sci. 2009, 122, 1939–1946.
- Peppelenbosch, M.; Boone, E.; Jones, G.E.; van Deventer, S.J.; Haegeman, G.; Fiers, W.; Grooten, J.; Ridley, A.J. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: Inhibition of Cdc42-mediated filopodium formation by TNF. J. Immunol. 1999, 162, 837–845.
- Puls, A.; Eliopoulos, A.G.; Nobes, C.D.; Bridges, T.; Young, L.S.; Hall, A. Activation of the small GTPase Cdc42 by the inflammatory cytokines TNF(alpha) and IL-1, and by the Epstein-Barr virus transforming protein LMP1. J. Cell Sci. 1999, 112 Pt 17, 2983–2992.
- Murali, A.; Rajalingam, K. Small Rho GTPases in the control of cell shape and mobility. Cell. Mol. Life Sci. CMLS 2014, 71, 1703–1721.
- Geisbrecht, E.R.; Montell, D.J. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell 2004, 118, 111–125.
- Marivin, A.; Berthelet, J.; Cartier, J.; Paul, C.; Gemble, S.; Morizot, A.; Boireau, W.; Saleh, M.; Bertoglio, J.; Solary, E.; et al. cIAP1 regulates TNF-mediated cdc42 activation and filopodia formation. Oncogene 2014, 33, 5534–5545.
- Oberoi, T.K.; Dogan, T.; Hocking, J.C.; Scholz, R.P.; Mooz, J.; Anderson, C.L.; Karreman, C.; Meyer Zu Heringdorf, D.; Schmidt, G.; Ruonala, M.; et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2011, 31, 14–28.
- Oberoi-Khanuja, T.K.; Rajalingam, K. Ubiquitination of Rac1 by inhibitors of apoptosis (IAPs). Methods Mol. Biol. 2014, 1120, 43–54.
- Hornburger, M.C.; Mayer, B.A.; Leonhardt, S.; Willer, E.A.; Zahler, S.; Beyerle, A.; Rajalingam, K.; Vollmar, A.M.; Furst, R. A novel role for inhibitor of apoptosis (IAP) proteins as regulators of endothelial barrier function by mediating RhoA activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1938–1946.
- Murali, A.; Shin, J.; Yurugi, H.; Krishnan, A.; Akutsu, M.; Carpy, A.; Macek, B.; Rajalingam, K. Ubiquitin-dependent regulation of Cdc42 by XIAP. Cell Death Dis. 2017, 8, e2900.
- Dubrez, L.; Rajalingam, K. IAPs and cell migration. Semin. Cell Dev. Biol. 2015, 39, 124–131.
- Sweeney, M.C.; Wang, X.; Park, J.; Liu, Y.; Pei, D. Determination of the sequence specificity of XIAP BIR domains by screening a combinatorial peptide library. Biochemistry 2006, 45, 14740–14748.
- He, W.; Wang, Q.; Srinivasan, B.; Xu, J.; Padilla, M.T.; Li, Z.; Wang, X.; Liu, Y.; Gou, X.; Shen, H.M.; et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 2014, 33, 3004–3013.
- Lee, S.; Lee, J.Y.; Lee, E.W.; Park, S.; Kang, D.H.; Min, C.; Lee, D.J.; Kang, D.; Song, J.; Kwon, J.; et al. Absence of Cytosolic 2-Cys Prx Subtypes I and II Exacerbates TNF-α-Induced Apoptosis via Different Routes. Cell Rep. 2019, 26, 2194–2211.e2196.
- Romagny, S.; Bouaouiche, S.; Lucchi, G.; Ducoroy, P.; Bertoldo, J.B.; Terenzi, H.; Bettaieb, A.; Plenchette, S. S-Nitrosylation of cIAP1 Switches Cancer Cell Fate from TNFα/TNFR1-Mediated Cell Survival to Cell Death. Cancer Res. 2018, 78, 1948–1957.

