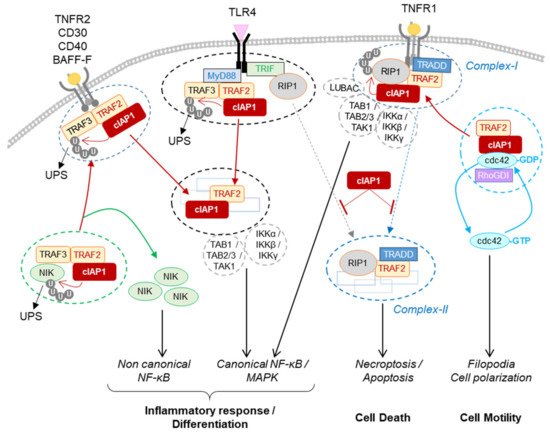
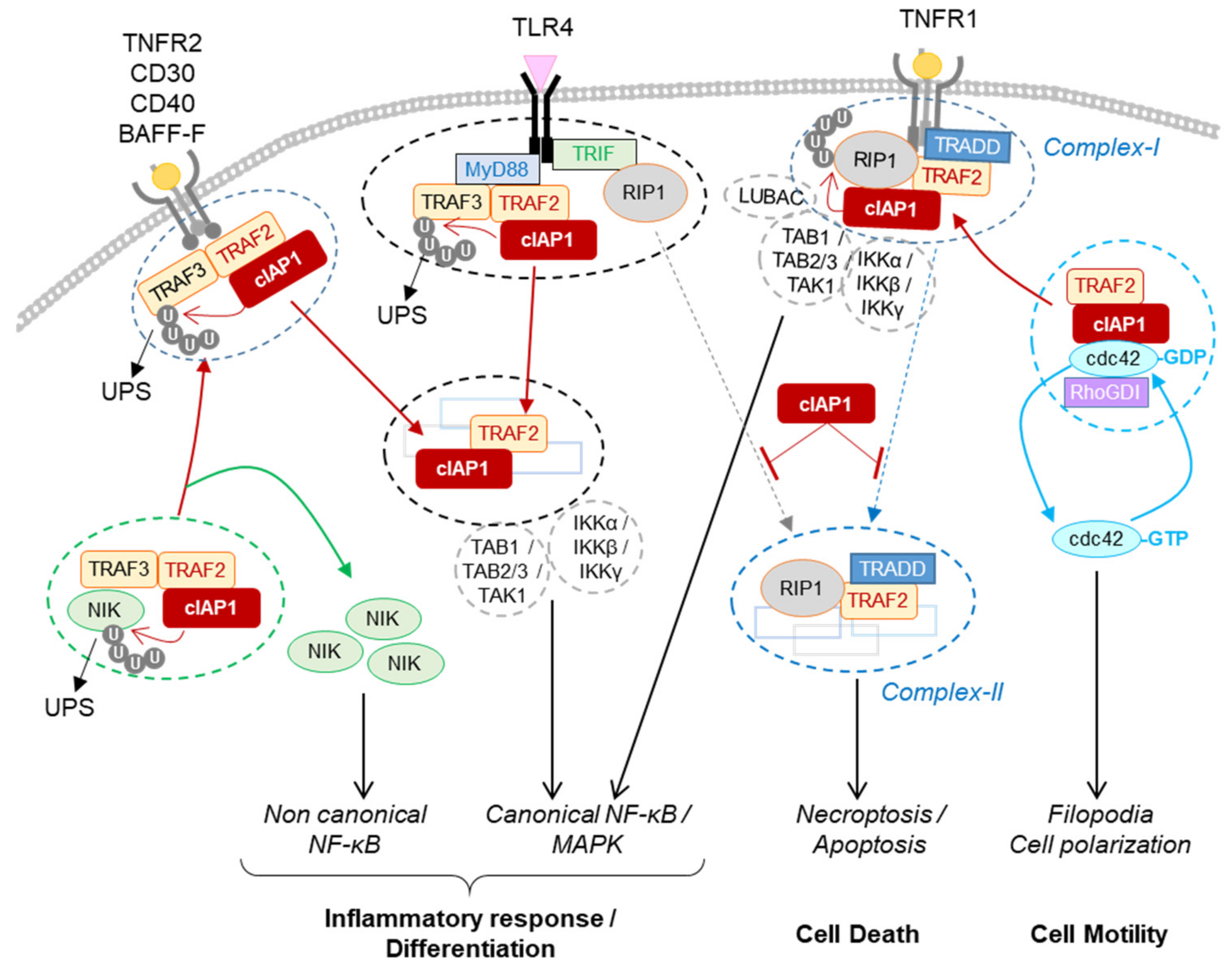
Cell shape and cell motility are controlled by small GTPases of the Rho family. These proteins are critical regulators of the dynamic reorganization of the actin cytoskeleton
[74][75][76][101,102,103]. They control cell architecture, focal adhesion complexes and local contraction by promoting the generation of stress fibers or membrane protrusions such as lamellopodia or filopodia
[77][104]. They switch between a cytoplasmic, inactive GDP-bound state and a membrane-associated, active GTP-bound state, providing energy required for cytoskeleton rearrangement. RhoGTPase activation is mediated by guanine-nucleotide exchange factors (GEFs), which catalyze the transfer of GDP-bound to GTP-bound forms. Once activated, RhoGTPases are either recycled in inactive state by the action of GTPase-activation proteins (GAPs) or subjected to UPS-mediated degradation. The activation cycle of Rho GTPase is controlled by molecular chaperones such as guanine-nucleotide dissociation inhibitors (GDIs) which stabilize Rho GTPases in their cytosolic inactive state
[77][104]. A relationship between IAPs and RhoGTPases was suggested in 2004 in a study showing that drosophila DIAP1 can interact with Rac1 and compensate for the migration defect triggered by the expression of a dominant negative form of this GTPase
[78][105]. In mammals, in vitro assays have demonstrated that cIAP1, cIAP2 and XIAP are able to directly interact with the three most studied RhoGTPases
[79][80][81][82][106,107,108,109] RhoA, Rac1 and cdc42, which promote lamellopodia, stress fibers or filopidia, respectively. In a study analysing the influence of cIAP1 on cell shape and migration,
it'swe demonstrated that cIAP1 can directly bind cdc42. It stabilizes cdc42 in its GDP-, inactive-state by promoting its association with its molecular chaperone RhoGDI. Deletion of cIAP1 deregulated the activation cycle of cdc42 by promoting its activation and then degradation
[79][106]. Accordingly, cIAP1
−/− fibroblasts display an enhanced ability to migrate and exhibit filopodia. TNFα has the ability to induce cdc42 activation and actin reorganisation
[75][76][102,103]. Upon TNFα stimulation, cIAP1 is recruited to the membrane receptor-associated complex, releasing cdc42 and promoting its activation
[79][106] (
Figure 14). The ubiquitination of cdc42 by cIAP1 has not been demonstrated; however, the ability of XIAP to ubiquitinate cdc42 and of XIAP and cIAP1 to ubiquitinate Rac1 has been observed
[80][83][107,110]. Single or combined deletion of cIAP1, cIAP2 or XIAP differently affects cell shape, actin distribution and migratory capacity. They appear to have specific and distinct activity on each of the Rho proteins, suggesting that IAPs could regulate the spatiotemporal and sequential activation of Rho proteins
[84][111]. Additional analysis will be required to decipher the regulation of the Rho proteins by IAPs.
3. Conclusions
cIAP1 mainly exerts its activity by controlling the cell fate of its protein partners. Thanks to their ability to promote the conjugation of ubiquitin chains of different types, they can modulate the stability, localization and/or the activity of intracellular proteins and can change the composition of signaling platforms by modifying the intermolecular binding affinities. Thus, IAPs have the ability to control the implementation of signaling pathways and their regulations in time and space. To date, more than 30 cIAP substrates have been identified (recently reviewed in
[15]). A database search for proteins containing IBM-like sequences found many proteins with different cellular functions
[85][133], greatly expanding the number of potential IAP-binding partners. The identified IAP substrates are involved in various cellular processes essential for maintaining cell homeostasis (innate immune response, DNA damage response, cell cycle regulation). For most of them, the type and site of ubiquitination have not been determined. However, this is an important issue to address since they determine the cellular fate of the substrate
[48][43].
The ultimate function of IAPs is to allow cells to adapt to their changing environment, to help implement an appropriate response to combat endogenous or exogenous stress or microbial aggression, and to restore homeostasis. Although loss of cIAP1 in mice has been associated with locale inflammation in lung, intestines or skin
[45][46][78,79], deletion or mutation of the BIRC2/3 gene has not been associated with chronic inflammatory disease but has with cancer development. More in-depth studies of the implication of cIAPs in these pathologies deserve to be carried out. Most studies have focused on analyzing the role of cIAPs in innate immunity and in regulating cell surface receptor signaling pathways. However, consistent with their nuclear expression in cells in many tissues, their functions in the nucleus, in particular as a transcriptional regulator, may have been underestimated.
The expression of cIAP1 is ubiquitous and its regulation mechanisms are still poorly understood. The last observations suggest that cIAP1 and TRAF2 require each other and form an E3-ubiquitin ligase complex. cIAP1 E3-ligase activity is stimulated by K63-linked ubiquitination that can be mediated by TRAF2 or TRAF6
[55][70][34,98]. The stability of cIAP1 can be controlled by phosphorylation
[86][114], and its regulation by S-nitrosylation and oxidation processes have also been reported
[87][88][115,116]. One important issue to address concerns the mechanisms of regulation of the subcellular localization of cIAP1.
Smac mimetics designed to block XIAP anti-apoptotic activity are also potent inhibitors of cIAP1 by promoting its proteasome-mediated degradation. They have been developed as anticancer agents. However, because of the ability of cIAP1 to regulate RIP1 activities, numerous preclinical studies are exploring their potential in the treatment of inflammatory and infectious disease
.