Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Angela Rocca | + 2983 word(s) | 2983 | 2022-03-08 04:08:40 | | | |
| 2 | Catherine Yang | -1 word(s) | 2982 | 2022-03-11 02:03:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rocca, A. HFpEF. Encyclopedia. Available online: https://encyclopedia.pub/entry/20420 (accessed on 25 July 2026).
Rocca A. HFpEF. Encyclopedia. Available at: https://encyclopedia.pub/entry/20420. Accessed July 25, 2026.
Rocca, Angela. "HFpEF" Encyclopedia, https://encyclopedia.pub/entry/20420 (accessed July 25, 2026).
Rocca, A. (2022, March 10). HFpEF. In Encyclopedia. https://encyclopedia.pub/entry/20420
Rocca, Angela. "HFpEF." Encyclopedia. Web. 10 March, 2022.
Copy Citation
Heart failure (HF) with preserved left ventricular ejection fraction (HFpEF) is becoming the predominant form of HF. However, medical therapy that improves cardiovascular outcome in HF patients with almost normal and normal systolic left ventricular function, but diastolic dysfunction is missing. The cause of this unmet need is incomplete understanding of HFpEF pathophysiology, the heterogeneity of the patient population, and poor matching of therapeutic mechanisms and primary pathophysiological processes.
heart failure with preserved left ventricular ejection fraction
cardiomyocyte
1. Structural and Functional Changes of the Cardiomyocyte in HFpEF
Independent of this diagnostic dilemma, enormous progress has been made in the comprehension of HFpEF disease in recent years with translational studies that investigated the origin of diastolic dysfunction. A remarkable study in the human investigated left ventricular epicardial anterior wall biopsy specimens obtained from patients with LVEF ≥ 50%, normal wall motion, and end-diastolic volume index < 75 mL/m2 undergoing coronary artery bypass grafting [1]. Patients were divided into groups without hypertension (HTN) (control group), with HTN but without HFpEF, and HTN with HFpEF. Compared to controls, patients with HTN but without HFpEF had no change in LVEDP, myocardial passive stiffness, collagen, or titin phosphorylation, but showed an increase of serum biomarkers of inflammation (C-reactive protein, ST2, tissue inhibitor of metalloproteinase-1). However, patients with HTN and HFpEF had high LVEDP, increased left atrial volume, and elevated levels of NT-proBNP or biomarkers of inflammation. Most importantly, overall stiffness was increased with collagen and titin contributing simultaneously based on correlation coefficient analysis. In summary, these changes suggest that HFpEF involves changes in collagen and titin homeostasis and that proinflammatory and profibrotic stimuli may play a role [1].
In fact, the giant cytoskeletal protein titin, which is the largest known protein [2], acts as a bidirectional spring and is responsible for early diastolic recoil and late diastolic distensibility of cardiomyocytes. It exists in two cardiac titin isoforms: the more compliant N2BA and the stiffer N2B isoform [3][4] and the N2BA:N2B ratio defines myofibrillar passive stiffness [5][6][7][8]. Of note, in a murine model of HFpEF there is an increase in the expression of the two isoforms with a relative reduction of the N2BA [9]. However, LV stiffness also depends on the phosphorylation state of titin, and, in HFpEF patients when compared to controls titin phosphorylation on PEVK S11878 (S26) (the proline, glutamate, valine, and lysine domain) is more important, while phosphorylation on N2B S4185(S469) is decreased [10]. The phosphorylation state of titin is modulated by different protein kinases: the cAMP-dependent protein kinase (PKA), the cGMP-dependent protein kinase (PKG), and the protein kinase C (PKC) [11]. Phosphorylation by PKA or PKG render titin more compliant, while protein-kinase-C-dependent phosphorylation decreases compliance [12]. In detail, phosphorylation of N2B (N2Bus segment) reduces cardiomyocyte resting tension (Fpassive), while PKC mediated phosphorylation at the PEVK domain increases stiffness [13][14]. Therefore, a deficit in phosphorylation at the N2Bus-titin site by PKA/PKG or an increased phosphorylation at the PEVK domain by PKC leads to more important Fpassive and thus stiffness [13][14]. PKG activity is in general decreased in HFpEF, while PKC is elevated, resulting in imbalanced phosphorylation and increased diastolic stiffness [10][15][16][17] (Figure 1). This can explain why sildenafil reduces diastolic stiffness in an old hypertensive dog model and also in HFpEF patients on chronic sildenafil treatment for pulmonary hypertension [3].
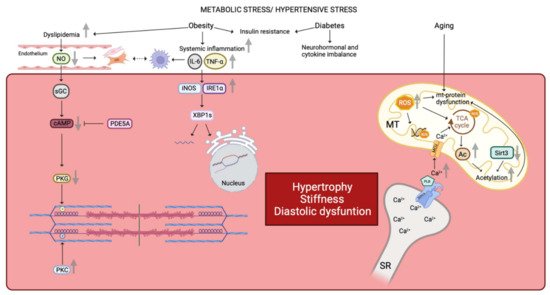
Figure 1. Similarities in human and animal HFpEF pathophysiology. Metabolic stress and hypertensive stress impaired cardiomyocyte functionality. Obesity favors dyslipidemia, which leads to NO reduction and finally a reduced PKG activity by the NO-sGC-cAMP pathway. Reduced PKG activity is correlated with a decreased N2B phosphorylation. On the contrary, PKC activity is increased in HFpEF disease leading to an increased phosphorylation state of the titin’s PEVK domain. Moreover, reduced NO bioavailability promotes fibroblasts and myofibroblasts proliferation. These latter interact with infiltrated macrophages which are linked with the inflammatory state. Indeed, obesity favors systemic inflammation with an increased concentrations of IL-6 and TNF-α. Increased plasma levels of Il-6 and TNF-α impair gene expression as well as the correct folding of proteins by the iNOS-IRE1α-XBP1 pathway. Both obesity and diabetes comorbidities are linked with insulin resistance, neurohormonal and cytokine imbalance affecting the cardiomyocyte environment. Finally, aging alters mitochondrial function by increased ROS production. ROS impact on mitochondrial DNA, mitochondrial proteins, and TCA cycle. Impairment of TCA cycle is also due to the increased mitochondrial Ca2+ concentration. Impairment of the TCA cycle leads to an increased Acetyl-CoA concentration, due to a decreased expression of mitochondrial deacetylase Sirt3, and results in hyperacetylation of mitochondrial proteins. Furthermore, increased mitochondrial Ca2+ concentration results in increased PLB activity, which inhibits SERCA and promotes cytosolic Ca2+ accumulation, which passes into the mitochondria via the MCU. Altogether, these pathways lead to a deterioration of the cardiomyocyte state by promoting hypertrophy, stiffness, and diastolic dysfunction. NO: nitric oxide, PKG: protein kinase G, sGC: soluble guanylate cyclase, cAMP: cyclic adenosine monophosphate, PKC: protein kinase C, PEVK domain: proline-glutamate-valine-lysine domain, Il-6: interleukine 6, TNF-α: tumor necrosis factor α, iNOS: inducible nitric oxide synthase, IRE1α: inositol requiring endoribonuclease 1α, XBP1: X-box binding protein 1, ROS: reactive oxygen species, TCA: tricarboxylic acid, Ca2+: calcium, SERCA: sarcoplasmic/endoplasmic reticulum calcium ATPase, PLB: phospholamban, MCU: mitochondrial calcium uniporter, Ac: acetyl group, Sirt3: Sirtuin 3, SR: sarcoplasmic reticulum, MT: mitochondria, mt: mitochondrial, PDE5A: phosphodiesterase type 5A, P: group phosphate.
2. The Impact of Age, Hypertension, Obesity, and Diabetes in HFpEF
Systemic hypertension is the single most common comorbidity in HFpEF with a prevalence ranging from 60 to 96% in epidemiological studies, HF registries and large randomized controlled trials [18][19]. The relationship between hypertension and LV hypertrophy is well known since elevated blood pressure in midlife results in LV hypertrophy in later life [20]. Systemic hypertension furthermore induces arterial stiffness, which by itself results in a disproportionate afterload increase, up to the point of ventricular-vascular uncoupling [21]. Animal models such as the aldosterone-infused and unilateral nephrectomized mouse and the angiotensin-II infused mouse reproduced these characteristics. Both murine models develop hypertension, concentric LV hypertrophy, pulmonary congestion, and evidence of diastolic dysfunction and exertion intolerance while maintaining a normal or preserved LVEF. Beyond these typical clinical characteristics repeating human HFpEF, these mouse models reproducing other aspects of human HFpEF such as changes in titin NB2A and NB2B expression and an increase of extracellular matrix quantity [9][10][22]. This concordance suggests that animal models repeating chronic hypertension can serve for further translational studies in HFpEF. Results from epidemiological studies in humans indicate the importance of arterial hypertension since arterial hypertension was a key parameter in three of the five different clusters identified by latent class analysis of the SwedeHF study. In detail, arterial hypertension clustered with non-diabetic participants presenting with atrial fibrillation (30% of all cohort participants), obese patients (15%), and older participants with ischemic heart disease and renal dysfunction (20%) [23].
However, neither the rodent aging models nor the hypertension models contain aspects of the metabolic comorbidity in HFpEF, in particular obesity and diabetes. In fact, large outcome trials and registries reveal that being overweight or obese is a major risk factor of HFpEF [24]. There are multiple mechanisms whereby obesity contributes to HFpEF, since adiposity increases systemic inflammation, insulin resistance, and dyslipidemia, and impairs arterial, skeletal muscle, and physical function [25], all of which are abnormal in HFpEF [26] (Figure 1). In contrast, weight loss after bariatric surgery in the human decreases LV hypertrophy, and LV filling pressures and reduces diastolic dysfunction [27]. Furthermore, exercise capacity improves with caloric restriction and aerobic exercise training in human HFpEF [28]. HFpEF patients with morbid obesity (mean body mass index (BMI) 41 kg/m2) behave differently when compared to HFpEF patients with elevated systolic blood pressure and left ventricular hypertrophy. Their hemodynamic burden is less important, the systolic force-Ca2+ dependence is reduced by 50%, indicating depressed contractility, and passive stiffening is less important [29][30]. The molecular mechanisms for obesity-associated depression of sarcomere function remain largely unknown, but these findings call conventional views about HFpEF and cardiac contractility into question. Further support for a role of BMI in HFpEF derives from RNA sequencing of right ventricular septal endomyocardial biopsies of HFpEF patients. This study showed minimal overlap with gene expression in HFrEF and healthy donor controls in a principal component analysis. Upregulated genes in human HFpEF were enriched in mitochondrial adenosine triphosphate synthesis/electron transport, while these pathways were downregulated in HFrEF. Body mass index largely accounted for upregulated genes in HFpEF, whereas neither BMI nor other comorbidity was associated with pathways enriched in downregulated genes such as genes engaging endoplasmic reticulum stress, autophagy, and angiogenesis [31] (Figure 1). Yet, information on the effects of obesity alone from animal models is sparse since most animal models also present obesity with either insulin resistance or diabetes [4]. Thus, replication of this association of upregulated gene expression with BMI in an animal model is missing so far.
Diabetes and obesity are two components of the cardiometabolic syndrome, which comprises also arterial hypertension and dyslipidemia and the prevalence of diastolic dysfunction is 12–45% with increasing severity of obesity, 50% in pre-diabetes, and 70% in type 2 diabetes [32]. In particular, diabetes confers a significant additional increase of the relative risk for cardiovascular death and hospitalization in all HFpEF patients [33]. The major pathomechanisms involved with diabetes are cardiac insulin resistance as well as neurohormonal and cytokine imbalance. Insulin resistance results in insufficient energy supply of the cardiomyocyte with subsequent hypophosphorylation resulting in increased cardiomyocyte stiffness and impairment of diastolic function, whereas cytokine imbalance activates the mitogen-activated protein kinase (MAPK) signaling pathway, which again favors myocellular hypertrophy and cardiac fibrosis. [33] (Figure 1).
Most murine models of obesity and diabetes-induced HFpEF face the limitation that the rodent phenotype develops most often fulminant, while the human phenotype develops over years or even a longer time. One of the animal models that repeats slowly developing HFpEF is the db/db leptin-deficient mouse, which develops obesity concomitant with severe hyperglycemia due to type 2 diabetes. In early disease (eight weeks), animals show increase of inflammatory cytokines levels, whereas there is decrease at older age (28 weeks). Furthermore, LV hypertrophy develops only at older age along with enlarged cardiomyocytes, an increase of cardiac fibrosis and capillary rarefication. In addition, this mouse shows abnormal ventriculoarterial coupling due to decreased arterial compliance and increased LV stiffness [34], which repeats a central characteristic of human HFpEF (Figure 2). Heart rate reduction with ivabradine improved in this murine model vascular stiffness, LV contractility, and diastolic function [35], but this treatment was without effect on cardiovascular outcomes in HFpEF patients [36]. Again, this result may question the applicability of results obtained in rodent studies; however, the result also suggests that the drug was not provided to the phenotype subtype of HFpEF patients with potential benefit from If channel inhibition.
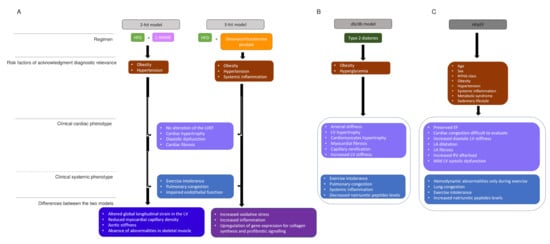
Figure 2. HFpEF models and human HFpEF diagrams. (A). Two-hit model versus three-hit model. two-hit model is based on exposition of mice to HFD and L-NAME to induce metabolic stress (obesity) and mechanical stress (hypertension). Obesity and hypertension are risk factors that underlie HFpEF. Three-hit model is induced by the treatment of mice with HFD and desoxycorticosterone pivalate, which lead to the same phenotypes as in the two-hit model and in addition present a systemic inflammation. The central boxes include the clinical cardiac phenotype and the clinical systemic phenotype present in both strategies. The two last boxes represents the differences observed in, respectively, two-hit and three-hit models. (B). The db/db model summarizes the phenotype observed in a murine model, which presents type 2 diabetes. (C). The human HFpEF diagram summarizes the clinical phenotype observed and HFpEF singularities. HDF: high fat diet, L-NAME: N-nitro-L-arginine methyl ester, LV: LV: left ventricule, LVEF: left ventricular ejection fraction, NYHA: New York heart association, EF: ejection fraction, LA: left atrial.
Notwithstanding of the role of hyperacetylization, the calcium metabolism may still play a role as suggested from a recent study investigating mitochondrial and cytosolic calcium handling in intact cardiomyocytes from ZSF1-obese animals [37][38]. Mitochondrial and cytosolic calcium level at rest and during contraction are in the ZSF1-obese rat higher when compared with lean ZSF1-controls. Myocardial excitation-contraction coupling begins by membrane depolarization that triggers Ca2+ entry via L-type calcium channels, which in turn stimulate release of Ca2+ ions from the sarcoplsmaic reticulum with subsequent myofilament activation and contraction with sarcomere shortening [39]. To terminate Ca2+-mediated myofilament interaction, the amount of cytosolic Ca2+ must decline which requires inactivation of L-type calcium channels and sequestration of cytosolic Ca2+ by the sarcoplasmic reticulum Ca−ATPase (SERCA2a) back into the sarcoplasmic reticulum. Relaxation is a key player of HFpEF pathophysiology, therefore, it is not surprising that modifications in proteins that mediate Ca2+ homeostasis have been reported. ZSF1-obese rats show higher cytosolic Ca2+ level resulting from a disbalanced interaction between phospholamban (PLB) and sarcoplasmic endoplasmic reticulum calcium pump (SERCA2a). This disbalance reduces the reuptake of Ca2+ ions via the SERCA2a back into the sarcoplasmic reticulum. In ZSF1-obese rats, the SERCA2a protein content was unchanged between groups when compared to the lean controls, while PLB expression was higher in the ZSF1 obese rat. Furthermore, hypophosphorylated PLB has also an inhibitory effect on SERCA2a activity, suggesting that disbalance of the PLB/SERCA2a expression ratio together with hypophosphorylation increase cytosolic Ca2+ levels in diastolic dysfunction. Since isolated hearts from rats with metabolic syndrome and diabetes have shown incapacity to upregulate ATP synthesis and decreased free energy of ATP hydrolysis (ΔG∼ATP) at higher workloads, exercise will worsen diastolic relaxation in addition [40][41]. Additional evidence for a role of calcium homeostasis in diastolic dysfunction derives from a study conducted in an aged murine model recapitulating age-related cardiac changes compatible with HFpEF. These animals demonstrated an association between SERCA2 protein expression and age-dependent diastolic dysfunction [42]. Coherent with this, gene expression of SERCA2a was reduced in human cardiac allografts with diastolic dysfunction resulting in a relative increase of PLB gene expression [43], while regulatory subunits of the L-type calcium channel decrease their gene expression with diastolic dysfunction in the human cardiac allograft most likely as a compensatory mechanism aiming to decrease inflow of exogeneous Ca2+ ions via the central pore of the cardial L-type calcium channel [44]. Taken together, these results indicate that slowed calcium reuptake into the sarcoplasmic reticulum [45] may explain increased cytosolic and mitochondrial higher calcium concentration in the ZSF1-obese rats, but also in humans (Figure 1).
However, increased cytosolic Ca2+ is not only associated with altered excitation-contraction coupling, but also with increased mitochondrial Ca2+. The latter is a regulator of NADH (complex 1) activity, which may explain the observation that mitochondrial respiration and oxidative phosphorylation are reduced in HFpEF [46][47][48][49]. Furthermore, sustained increase of mitochondrial calcium accumulation may also turn detrimental because it may promote opening of mitochondrial permeability transition pore, ultimately leading to cellular apoptosis [50]. In summary, these results suggest that change of mitochondrial function can ultimately result in bottlenecks of metabolic flux, redox imbalance, protein modification, ROS-induced ROS generation, impaired calcium homeostasis, and local inflammation [51] with the potential to affect cardiomyocyte function [52].
3. Impact of Systemic Inflammation on Cardiomyocyte Function, Cardiac Fibrosis, and Vascular Function
Cardiac fibrosis is defined as a state in which excess deposition of collagen in the extracellular matrix occurs. In the resting state of a healthy heart, fibroblasts constantly modify the extracellular environment, while an overall disproportionately increased extracellular matrix volume is a hallmark of HFpEF [1]. The importance of this pathological increase of extracellular matrix is highlighted by the fact several profibrotic markers (chitinase 3-like protein 1 (a marker of hepatic fibrosis), FGF23, IL6, MMP7, and sST2) were among the eight serum biomarkers that most accurately predict prognosis in study participants of the TOPCAT trial [53]. In coherence, the Karolinska Rennes biomarker study investigating 87 biomarkers and 240 clinical markers for their association with New York Heart Association (NYHA) class and the combined endpoint of all-cause mortality and HF hospitalization in participants with LVEF > 45% sorted growth/differentiation factor-15, a member of the TGF ß superfamily, as the strongest predictor after adjusting for age, sex, and N-terminal proBNP [54].
An increasingly popular idea about HFpEF is that systemic inflammation not only disrupts endothelial signaling, but also creates a stressful environment for the cardiomyocytes. In fact, an increased level of CD3+ cells and leucocytes positive for CD11a+, CD45+, and CD68nells was shown in right ventricular biopsy samples of HFpEF patients [55]. Further evidence for a role of immune cells in HFpEF derives from a study, which applied a broad T-cell inhibitor treatment in HFpEF patients resulting in slowed progression of cardiac hypertrophy and reduced activation and infiltration of T-cells and macrophages into the myocardium, and decreased loss of cardiomyocytes [56].
Last not least, systemic inflammation with the increased levels of pro-inflammatory mediators such as TNFα affects endothelial function of the coronary microvasculature with increased expression of endothelial adhesion molecules such as vascular cell adhesion molecule (VCAM) and E-selectin, as shown in myocardial biopsy samples from HFpEF patients [57][55][58]. Endothelial VCAM and E-selectin expression involve activation and the trans-subendothelial migration of circulating immune cells such as macrophages [55] into the myocardium due to vascular barriers weakening [59][55]. Macrophages infiltration has been demonstrated to be a key element of fibrosis through their interations with fibroblast leading to heart stiffness and myocardial dysfunction [60].
In parallel, endothelial cells are also affected by the systemic inflammation resulting in endothelial production of reactive oxygen species (ROS) due to activation of nicotinamide adenine dinucleotide phosphate oxidase [61]. This may also explain the high nitrosative/oxidative stress observed in HFpEF myocardium impacting on the mitochondrial function and thus the good working of cardiomyocytes [12][62][63]. Reduced NO bioavailability also promotes profibrotic action of the growth-promoting hormones endothelin-1, angiotensin II, and aldosterone favors collagen deposition [64], and more so in HFpEF, while less in HFrEF [65]. Furthermore, coronary microvascular endothelial inflammation leads to a reduced vasodilatation of the coronary microvascular bed as demonstrated by reduced acetylcholine-related vasodilation of the microcirculation in HFpEF [66]. This reduced response can explain left ventricular (LV) diastolic dysfunction [66] and a deficient systemic vasodilator response could also explain the reduced exercise tolerance [67]. However, this loss of exercise tolerance can be compensated thanks to exercise training program [68][69], which also to some extent improves diastolic LV dysfunction [68][69]. Indeed, there is evidence that either improvement is related to upregulation of endothelial nitric oxide synthase (eNOS) [68][69].
References
- Zile, M.R.; Baicu, C.F.; SIkonomidis, J.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial Stiffness in Patients with Heart Failure and a Preserved Ejection Fraction: Contributions of Collagen and Titin. Circulation 2015, 131, 1247–1259.
- Granzier, H.L.; Labeit, S. The Giant Protein Titin: A Major Player in Myocardial Mechanics, Signaling, and Disease. Circ. Res. 2004, 94, 284–295.
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. Pulmonary Hypertension in Heart Failure with Preserved Ejection Fraction: A target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011, 124, 164–174.
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2017, 2, 770–789.
- Freiburg, A.; Trombitas, K.; Hell, W.; Cazorla, O.; Fougerousse, F.; Centner, T.; Kolmerer, B.; Witt, C.; Beckmann, J.S.; Gregorio, C.C.; et al. Series of Exon-Skipping Events in the Elastic Spring Region of Titin as the Structural Basis for Myofibrillar Elastic Diversity. Circ. Res. 2000, 86, 1114–1121.
- Neagoe, C.; Kulke, M.; del Monte, F.; Gwathmey, J.K.; de Tombe, P.P.; Hajjar, R.J.; Linke, W.A. Titin Isoform Switch in Ischemic Human Heart Disease. Circulation 2002, 106, 1333–1341.
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.P.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered Titin Expression, Myocardial Stiffness, and Left Ventricular Function in Patients with Dilated Cardiomyopathy. Circulation 2004, 110, 155–162.
- Makarenko, I.; Opitz, C.A.; Leake, M.C.; Neagoe, C.; Kulke, M.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J.; Linke, W.A. Passive Stiffness Changes Caused by Upregulation of Compliant Titin Isoforms in Human Dilated Cardiomyopathy Hearts. Circ. Res. 2004, 95, 708–716.
- Valero-Munoz, M.; Li, S.; Wilson, R.M.; Boldbaatar, B.; Iglarz, M.; Sam, F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2016, 9, e003381.
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471.
- LeWinter, M.M.; Granzier, H. Cardiac Titin: A Multifunctional Giant. Circulation 2010, 121, 2137–2145.
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Borbély, A.; Falcao-Pires, I.; van Heerebeek, L.; Hamdani, N.; Édes, I.; Gavina, C.; Leite-Moreira, A.F.; Bronzwaer, J.G.F.; Papp, Z.; van der Velden, J.; et al. Hypophosphorylation of the Stiff N2B Titin Isoform Raises Cardiomyocyte Resting Tension in Failing Human Myocardium. Circ. Res. 2009, 104, 780–786.
- Hidalgo, C.; Hudson, B.; Bogomolovas, J.; Zhu, Y.; Anderson, B.; Greaser, M.; Labeit, S.; Granzier, H. PKC Phosphorylation of Titin’s PEVK Element: A Novel and Conserved Pathway for Modulating Myocardial Stiffness. Circ. Res. 2009, 105, 631–638.
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.V.; Bronzwaer, J.G.F.; van der Velden, J.; Stienen, G.J.M.; Laarman, G.J.; Somsen, A.; et al. Low Myocardial Protein Kinase G Activity in Heart Failure with Preserved Ejection Fraction. Circulation 2012, 126, 830–839.
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; de Tombe, P.P. Augmented Protein Kinase C-α–Induced Myofilament Protein Phosphorylation Contributes to Myofilament Dysfunction in Experimental Congestive Heart Failure. Circ. Res. 2007, 101, 195–204.
- Hudson, B.; Hidalgo, C.; Saripalli, C.; Granzier, H. Hyperphosphorylation of Mouse Cardiac Titin Contributes to Transverse Aortic Constriction-Induced Diastolic Dysfunction. Circ. Res. 2011, 109, 858–866.
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620.
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2013, 62, e147–e239.
- Ghosh, A.K.; Hardy, R.J.; Francis, D.P.; Chaturvedi, N.; Pellerin, D.; Deanfield, J.; Kuh, D.; Mayet, J.; Hughes, A.D. Midlife blood pressure change and left ventricular mass and remodelling in older age in the 1946 British birth cohort study. Eur. Heart J. 2014, 35, 3287–3295.
- Samson, R.; Jaiswal, A.; Ennezat, P.V.; Cassidy, M.; Le Jemtel, T.H. Clinical Phenotypes in Heart Failure With Preserved Ejection Fraction. JAHA 2016, 5, e002477.
- Tanaka, K.; Valero-Muñoz, M.; Wilson, R.M.; Essick, E.E.; Fowler, C.T.; Nakamura, K.; van den Hoff, M.; Ouchi, N.; Sam, F. Follistatin-Like 1 Regulates Hypertrophy in Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2016, 1, 207–221.
- Uijl, A.; Savarese, G.; Vaartjes, I.; Dahlström, U.; Brugts, J.J.; Linssen, G.C.M.; Empel, V.; Brunner-La Rocca, H.; Asselbergs, F.W.; Lund, L.H.; et al. Identification of distinct phenotypic clusters in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 973–982.
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90.
- Pandey, A.; Shah, S.J.; Butler, J.; Kellogg, D.L.; Lewis, G.D.; Forman, D.E.; Mentz, R.J.; Borlaug, B.A.; Simon, M.A.; Chirinos, J.A.; et al. Exercise Intolerance in Older Adults With Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2021, 78, 1166–1187.
- Kitzman, D.W.; Shah, S.J. The HFpEF Obesity Phenotype. J. Am. Coll. Cardiol. 2016, 68, 200–203.
- de las Fuentes, L.; Waggoner, A.D.; Mohammed, B.S.; Stein, R.I.; Miller, B.V.; Foster, G.D.; Wyatt, H.R.; Klein, S.; Davila-Roman, V.G. Effect of Moderate Diet-Induced Weight Loss and Weight Regain on Cardiovascular Structure and Function. J. Am. Coll. Cardiol. 2009, 54, 2376–2381.
- Kitzman, D.W.; Brubaker, P.; Morgan, T.; Haykowsky, M.; Hundley, G.; Kraus, W.E.; Eggebeen, J.; Nicklas, B.J. Effect of Caloric Restriction or Aerobic Exercise Training on Peak Oxygen Consumption and Quality of Life in Obese Older Patients With Heart Failure With Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA 2016, 315, 36.
- Aslam, M.I.; Hahn, V.S.; Jani, V.; Hsu, S.; Sharma, K.; Kass, D.A. Reduced Right Ventricular Sarcomere Contractility in Heart Failure With Preserved Ejection Fraction and Severe Obesity. Circulation 2021, 143, 965–967.
- Obokata, M.; Kane, G.C.; Reddy, Y.N.V.; Olson, T.P.; Melenovsky, V.; Borlaug, B.A. Role of Diastolic Stress Testing in the Evaluation for Heart Failure With Preserved Ejection Fraction: A Simultaneous Invasive-Echocardiographic Study. Circulation 2017, 135, 825–838.
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation 2021, 143, 120–134.
- Wan, S.-H.; Vogel, M.W.; Chen, H.H. Pre-Clinical Diastolic Dysfunction. J. Am. Coll. Cardiol. 2014, 63, 407–416.
- Kindermann, M.; Bohm, M. Sweet hearts die earlier--lessons from CHARM. Eur. Heart J. 2008, 29, 1342–1343.
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of Leptin Signaling Contributes to Cardiac Hypertrophy Independently of Body Weight in Mice. Circulation 2003, 108, 754–759.
- Reil, J.-C.; Hohl, M.; Reil, G.-H.; Granzier, H.L.; Kratz, M.T.; Kazakov, A.; Fries, P.; Müller, A.; Lenski, M.; Custodis, F.; et al. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur. Heart J. 2013, 34, 2839–2849.
- Komajda, M.; Isnard, R.; Cohen-Solal, A.; Metra, M.; Pieske, B.; Ponikowski, P.; Voors, A.A.; Dominjon, F.; Henon-Goburdhun, C.; Pannaux, M.; et al. Effect of ivabradine in patients with heart failure with preserved ejection fraction: The EDIFY randomized placebo-controlled trial: Ivabradine in HFpEF. Eur. J. Heart Fail. 2017, 19, 1495–1503.
- van den Brom, C.E.; Huisman, M.C.; Vlasblom, R.; Boontje, N.M.; Duijst, S.; Lubberink, M.; Molthoff, C.F.; Lammertsma, A.A.; van der Velden, J.; Boer, C.; et al. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: Studies using positron emission tomography. Cardiovasc. Diabetol. 2009, 8, 39.
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falcão-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; López, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial Titin Hypophosphorylation Importantly Contributes to Heart Failure with Preserved Ejection Fraction in a Rat Metabolic Risk Model. Circ. Heart Fail. 2013, 6, 1239–1249.
- Hullin, R.; Biel, M.; Flockerzi, V.; Hofmann, F. Tissue-specific expression of calcium channels. Trends Cardiovasc. Med. 1993, 3, 48–53.
- Tian, R.; Ingwall, J.S. Energetic basis for reduced contractile reserve in isolated rat hearts. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H1207–H1216.
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H192–H202.
- Dai, D.-F.; Rabinovitch, P.S. Cardiac Aging in Mice and Humans: The Role of Mitochondrial Oxidative Stress. Trends Cardiovasc. Med. 2009, 19, 213–220.
- Stüdeli, R.; Jung, S.; Mohacsi, P.; Perruchoud, S.; Castiglioni, P.; Wenaweser, P.; Heimbeck, G.; Feller, M.; Hullin, R. Diastolic Dysfunction in Human Cardiac Allografts is Related with Reduced SERCA2a Gene Expression: Reduced SERCA2a Expression in Diastolic Failure. Am. J. Transplant. 2006, 6, 775–782.
- Hullin, R.; Asmus, F.; Ludwig, A.; Hersel, J.; Boekstegers, P. Subunit Expression of the Cardiac L-Type Calcium Channel Is Differentially Regulated in Diastolic Heart Failure of the Cardiac Allograft. Circulation 1999, 100, 155–163.
- Miranda-Silva, D.; Wüst, R.C.I.; Conceição, G.; Gonçalves-Rodrigues, P.; Gonçalves, N.; Gonçalves, A.; Kuster, D.W.D.; Leite-Moreira, A.F.; Velden, J.; Sousa Beleza, J.M.; et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. 2020, 228, e13378.
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007, 102, 369–392.
- Liu, T.; O’Rourke, B. Enhancing Mitochondrial Ca2+ Uptake in Myocytes From Failing Hearts Restores Energy Supply and Demand Matching. Circ. Res. 2008, 103, 279–288.
- Dorn, G.W.; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49.
- Wüst, R.C.I.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.M.; Stienen, G.J.M. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016, 111, 362–372.
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394.
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726.
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421.
- Chirinos, J.A.; Orlenko, A.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Spires, T.E.; Yarde, M.; Wang, Z.; Seiffert, D.A.; et al. Multiple Plasma Biomarkers for Risk Stratification in Patients With Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 75, 1281–1295.
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.-C.; Gan, L.-M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure with Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10, e001633.
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac Inflammation Contributes to Changes in the Extracellular Matrix in Patients with Heart Failure and Normal Ejection Fraction. Circ. Heart Fail. 2011, 4, 44–52.
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Ormbostad Berre, A.M.; Stølen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680.
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.J.; Schalkwijk, C.G.; Bronzwaer, J.G.F.; Diamant, M.; et al. Diastolic Stiffness of the Failing Diabetic Heart: Importance of Fibrosis, Advanced Glycation End Products, and Myocyte Resting Tension. Circulation 2008, 117, 43–51.
- Cuijpers, I.; Simmonds, S.J.; van Bilsen, M.; Czarnowska, E.; González Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res. Cardiol. 2020, 115, 39.
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724.
- Falkenham, A.; de Antueno, R.; Rosin, N.; Betsch, D.; Lee, T.D.G.; Duncan, R.; Légaré, J. Nonclassical Resident Macrophages Are Important Determinants in the Development of Myocardial Fibrosis. Am. J. Pathol. 2015, 185, 927–942.
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501.
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453.
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.-P.; Ming, X.-F.; Yang, Z. Hyperactive S6K1 Mediates Oxidative Stress and Endothelial Dysfunction in Aging: Inhibition by Resveratrol. PLoS ONE 2011, 6, e19237.
- Zannad, F.; Radauceanu, A. Effect of MR Blockade on Collagen Formation and Cardiovascular Disease with a Specific Emphasis on Heart Failure. Heart Fail. Rev. 2005, 10, 71–78.
- Duprez, D.A.; Gross, M.D.; Kizer, J.R.; Ix, J.H.; Hundley, W.G.; Jacobs, D.R. Predictive Value of Collagen Biomarkers for Heart Failure With and Without Preserved Ejection Fraction: MESA (Multi-Ethnic Study of Atherosclerosis). JAHA 2018, 7, e007885.
- Tschöpe, C.; Bock, C.-T.; Kasner, M.; Noutsias, M.; Westermann, D.; Schwimmbeck, P.-L.; Pauschinger, M.; Poller, W.-C.; Kühl, U.; Kandolf, R.; et al. High Prevalence of Cardiac Parvovirus B19 Infection in Patients with Isolated Left Ventricular Diastolic Dysfunction. Circulation 2005, 111, 879–886.
- Borlaug, B.A.; Olson, T.P.; Lam, C.S.P.; Flood, K.S.; Lerman, A.; Johnson, B.D.; Redfield, M.M. Global Cardiovascular Reserve Dysfunction in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2010, 56, 845–854.
- Haykowsky, M.J.; Brubaker, P.H.; Stewart, K.P.; Morgan, T.M.; Eggebeen, J.; Kitzman, D.W. Effect of Endurance Training on the Determinants of Peak Exercise Oxygen Consumption in Elderly Patients with Stable Compensated Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2012, 60, 120–128.
- Edelmann, F.; Gelbrich, G.; Düngen, H.-D.; Fröhling, S.; Wachter, R.; Stahrenberg, R.; Binder, L.; Töpper, A.; Lashki, D.J.; Schwarz, S.; et al. Exercise Training Improves Exercise Capacity and Diastolic Function in Patients with Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2011, 58, 1780–1791.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
11 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No