 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammed Abdullah Alshawsh | + 11085 word(s) | 11085 | 2022-03-01 03:53:10 | | | |
| 2 | Bruce Ren | Meta information modification | 11085 | 2022-03-07 09:06:13 | | | | |
| 3 | Bruce Ren | Meta information modification | 11085 | 2022-03-07 09:07:06 | | |
Video Upload Options
Cancer is a life-threatening disease and is considered to be among the leading causes of death worldwide. Chemoresistance, severe toxicity, relapse and metastasis are the major obstacles in cancer therapy. Therefore, introducing new therapeutic agents for cancer remains a priority to increase the range of effective treatments. Terpenoids, a large group of secondary metabolites, are derived from plant sources and are composed of several isoprene units. The high diversity of terpenoids has drawn attention to their potential anticancer and pharmacological activities. Some terpenoids exhibit an anticancer effect by triggering various stages of cancer progression, for example, suppressing the early stage of tumorigenesis via induction of cell cycle arrest, inhibiting cancer cell differentiation and activating apoptosis. At the late stage of cancer development, certain terpenoids are able to inhibit angiogenesis and metastasis via modulation of different intracellular signaling pathways. Significant progress in the identification of the mechanism of action and signaling pathways through which terpenoids exert their anticancer effects has been highlighted.
1. Introduction
2. Mechanism of Action of Selected Terpenoids against Cancer Progression
2.1. Monoterpenoids
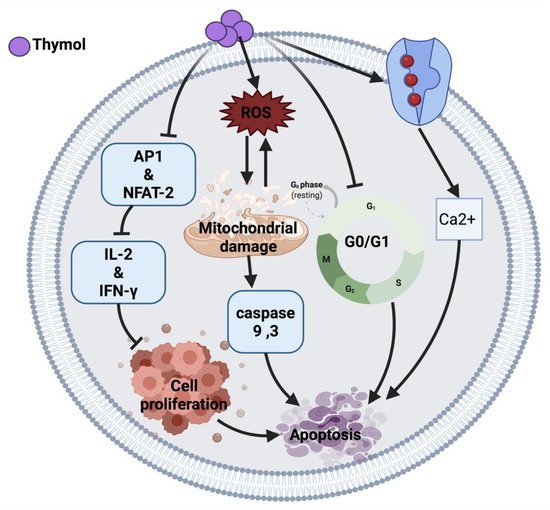
2.1.1. Thymol
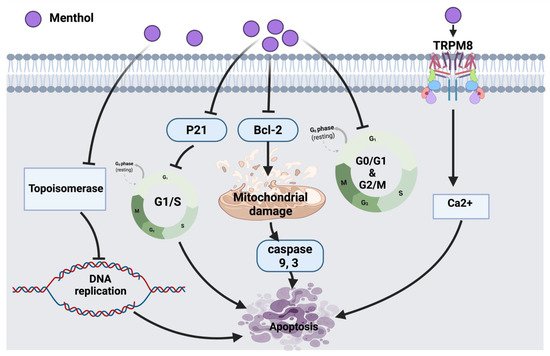
2.1.2. Menthol
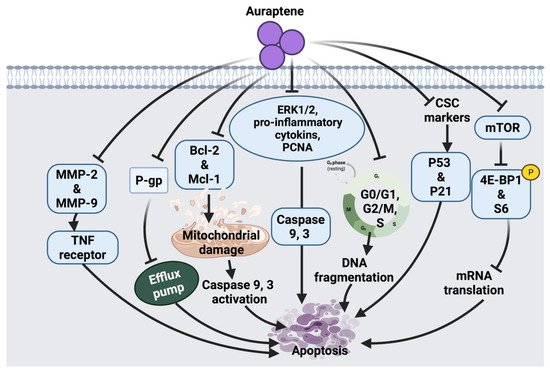
2.1.3. Auraptene
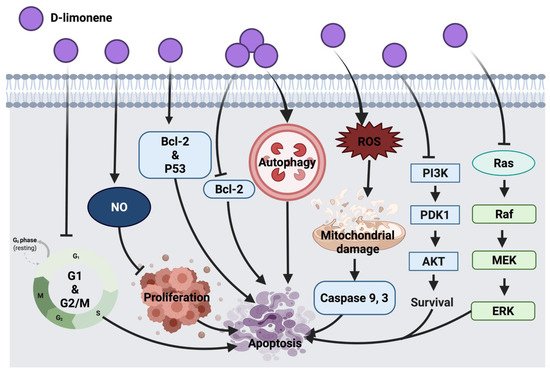
2.1.4. D-Limonene
2.1.5. Perillic Acid
2.1.6. Ascaridole
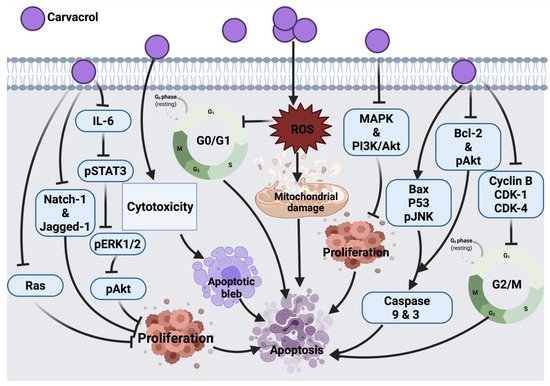
2.1.7. Carvacrol
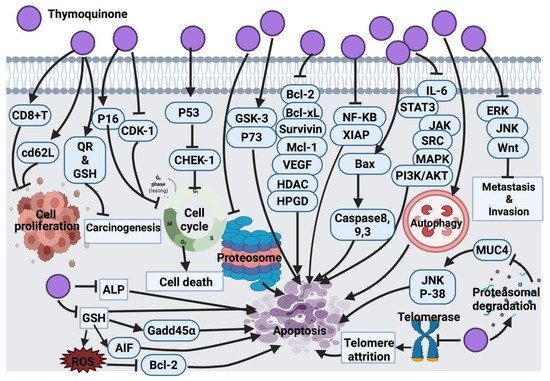
2.1.8. Thymoquinone
2.2. Sesquiterpenoids and Sesquiterpene Lactones
2.2.1. Sesquiterpene Lactones
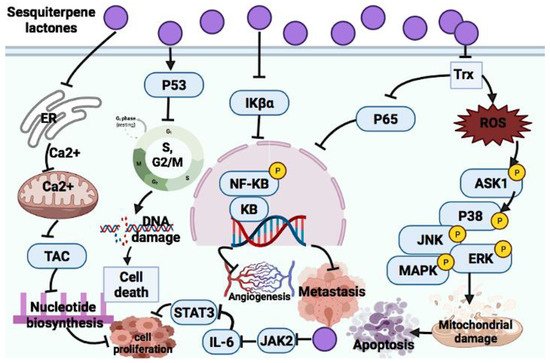
Parthenolide
Costunolide
Dehydrocostus Lactone
Helenalin
Elephantopus Mollis 23 (EM23)
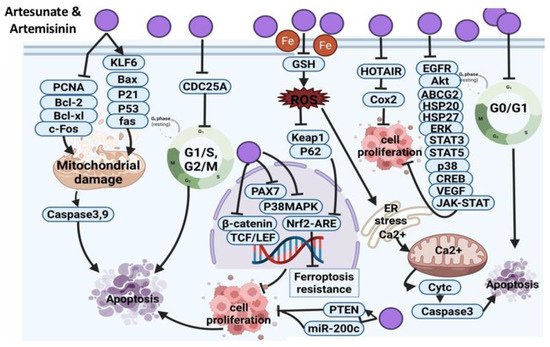
Artesunate and Artemisinin
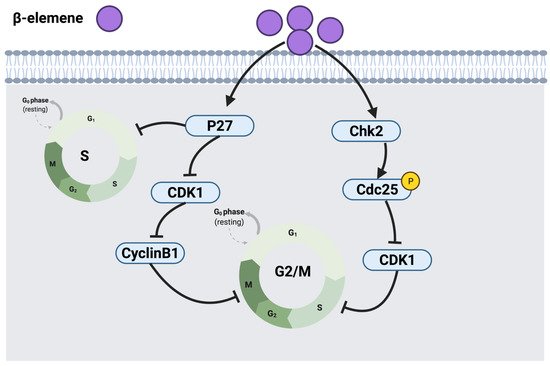
2.2.2. β-Elemene
2.3. Diterpenoids
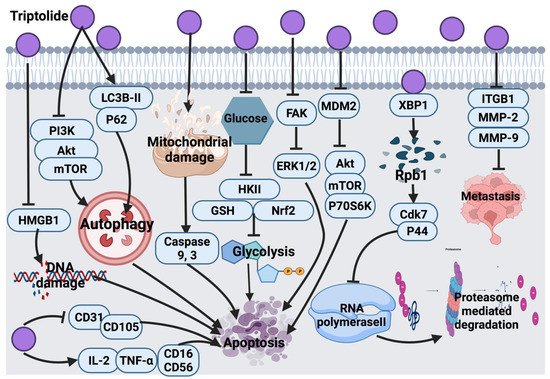
2.3.1. Triptolide
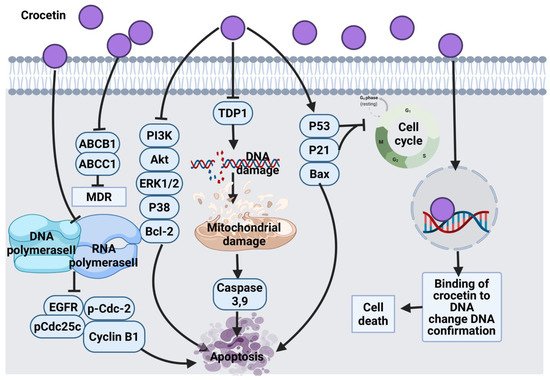
2.3.2. Crocetin
2.3.3. Phytol
2.4. Triterpenoids
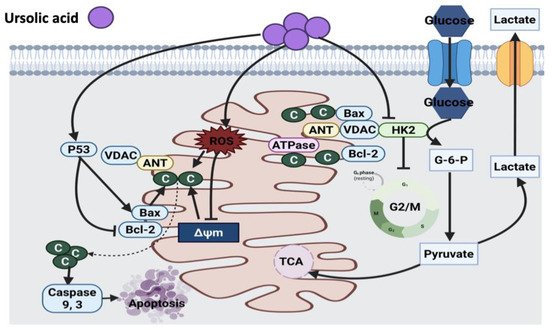
2.4.1. Ursolic Acid
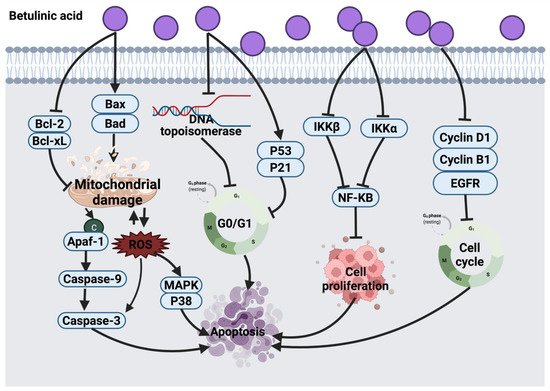
2.4.2. Betulinic Acid
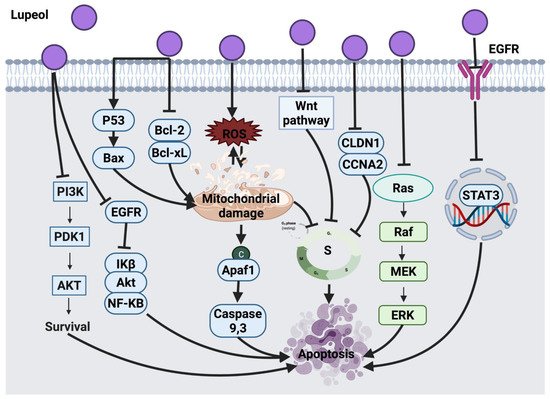
2.4.3. Lupeol
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248.
- Kondrashov, F.A.; Kondrashov, A.S. Measurements of spontaneous rates of mutations in the recent past and the near future. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1169–1176.
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157.
- Nevzorova, Y.A.; Grossmann, J.; Trautwein, C. Anti-tumorigenic and anti-angiogenic effects of natural conifer Abies sibirica terpenoids in vivo and in vitro. Biomed. Pharmacother. 2017, 89, 386–395.
- Li, D.; Du, Z.; Li, C.; Liu, Y.; Goodin, S.; Huang, H.; He, Y.; Zhang, Y.; Wang, H.; Zheng, X.; et al. Potent inhibitory effect of terpenoids from Acanthopanax trifoliatus on growth of PC-3 prostate cancer cells in vitro and in vivo is associated with suppression of NF-κB and STAT3 signalling. J. Funct. Foods 2015, 15, 274–283.
- Kuete, V.; Omosa, L.K.; Midiwo, J.O.; Karaosmanoğlu, O.; Sivas, H. Cytotoxicity of naturally occurring phenolics and terpenoids from Kenyan flora towards human carcinoma cells. J. Ayurveda Integr. Med. 2019, 10, 178–184.
- Deb, D.D.; Parimala, G.; Devi, S.S.; Chakraborty, T. Effect of thymol on peripheral blood mononuclear cell PBMC and acute promyelotic cancer cell line HL-60. Chem. Biol. Interact. 2011, 193, 97–106.
- Llana-Ruiz-Cabello, M.; Gutiérrez-Praena, D.; Pichardo, S.; Moreno, F.J.; Bermudez, J.M.; Aucejo, S.; Cameán, A.M. Cytotoxicity and morphological effects induced by carvacrol and thymol on the human cell line Caco-2. Food Chem. Toxicol. 2014, 64, 281–290.
- Kang, S.-H.; Kim, Y.-S.; Kim, E.-K.; Hwang, J.-W.; Jeong, J.-H.; Dong, X.; Lee, J.-W.; Moon, S.-H.; Jeon, B.-T.; Park, P.-J. Anticancer Effect of Thymol on AGS Human Gastric Carcinoma Cells. J. Microbiol. Biotechnol. 2016, 26, 28–37.
- Li, Y.; Wen, J.-M.; Du, C.-J.; Hu, S.-M.; Chen, J.-X.; Zhang, S.-G.; Zhang, N.; Gao, F.; Li, S.-J.; Mao, X.-W.; et al. Thymol inhibits bladder cancer cell proliferation via inducing cell cycle arrest and apoptosis. Biochem. Biophys. Res. Commun. 2017, 491, 530–536.
- Meeran, M.F.N.; Jagadeesh, G.S.; Selvaraj, P. Thymol attenuates altered lipid metabolism in β-adrenergic agonist induced myocardial infarcted rats by inhibiting tachycardia, altered electrocardiogram, apoptosis and cardiac hypertrophy. J. Funct. Foods 2015, 14, 51–62.
- De La Chapa, J.J.; Singha, P.K.; Lee, D.R.; Gonzales, C.B. Thymol inhibits oral squamous cell carcinoma growth via mitochondria-mediated apoptosis. J. Oral Pathol. Med. 2018, 47, 674–682.
- Gholijani, N.; Gharagozloo, M.; Kalantar, F.; Ramezani, A.; Amirghofran, Z. Modulation of Cytokine Production and Transcription Factors Activities in Human Jurkat T Cells by Thymol and Carvacrol. Adv. Pharm. Bull. 2015, 5, 653–660.
- Chang, H.-T.; Hsu, S.-S.; Chou, C.-T.; Cheng, J.-S.; Wang, J.-L.; Lin, K.-L.; Fang, Y.-C.; Chen, W.-C.; Chien, J.-M.; Lu, T.; et al. Effect of Thymol on Ca2+ Homeostasis and Viability in MG63 Human Osteosarcoma Cells. Pharmacology 2011, 88, 201–212.
- Hsu, S.-S.; Lin, K.-L.; Chou, C.-T.; Chiang, A.-J.; Liang, W.-Z.; Chang, H.-T.; Tsai, J.-Y.; Liao, W.-C.; Huang, F.-D.; Huang, J.K.; et al. Effect of thymol on Ca2+ homeostasis and viability in human glioblastoma cells. Eur. J. Pharmacol. 2011, 670, 85–91.
- Yeh, J.-H.; Chou, C.-T.; Chen, I.-S.; Lu, T.; Lin, K.-L.; Yu, C.-C.; Liang, W.-Z.; Chang, H.-T.; Kuo, C.-C.; Ho, C.-M.; et al. Effect of Thymol on Ca2+ Homeostasis and Viability in PC3 Human Prostate Cancer Cells. Chin. J. Physiol. 2017, 60, 32–40.
- Lin, J.-P.; Lu, H.-F.; Lee, J.-H.; Lin, J.-G.; Hsia, T.-C.; Wu, L.-T.; Chung, J.-G. (-)-Menthol inhibits DNA topoisomerases I, II alpha and beta and promotes NF-kappaB expression in human gastric cancer SNU-5 cells. Anticancer Res. 2005, 25, 2069–2074.
- Park, E.-J.; Kim, S.-H.; Kim, B.-J.; Kim, S.-Y.; So, I.; Jeon, J.-H. Menthol Enhances an Antiproliferative Activity of 1α,25-Dihydroxyvitamin D3 in LNCaP Cells. J. Clin. Biochem. Nutr. 2009, 44, 125–130.
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530.
- Li, Q.; Wang, X.; Yang, Z.; Wang, B.; Li, S. Menthol Induces Cell Death via the TRPM8 Channel in the Human Bladder Cancer Cell Line T24. Oncology 2009, 77, 335–341.
- Wang, Y.; Wang, X.; Yang, Z.; Zhu, G.; Chen, D.; Meng, Z. Menthol Inhibits the Proliferation and Motility of Prostate Cancer DU145 Cells. Pathol. Oncol. Res. 2012, 18, 903–910.
- Kim, S.-H.; Lee, S.; Piccolo, S.R.; Allen-Brady, K.; Park, E.-J.; Chun, J.N.; Kim, T.W.; Cho, N.-H.; Kim, I.-G.; So, I.; et al. Menthol induces cell-cycle arrest in PC-3 cells by down-regulating G2/M genes, including polo-like kinase 1. Biochem. Biophys. Res. Commun. 2012, 422, 436–441.
- Moon, J.Y.; Kim, H.; Cho, S.K. Auraptene, a Major Compound of Supercritical Fluid Extract of Phalsak (CitrusHassaku Hort ex Tanaka), Induces Apoptosis through the Suppression of mTOR Pathways in Human Gastric Cancer SNU-1 Cells. Evid. Based Complement. Altern. Med. 2015, 2015, 402385.
- Saboor-Maleki, S.; Rassouli, F.B.; Matin, M.M.; Iranshahi, M. Auraptene Attenuates Malignant Properties of Esophageal Stem-Like Cancer Cells. Technol. Cancer Res. Treat. 2017, 16, 519–527.
- Jamialahmadi, K.; Salari, S.; Alamolhodaei, N.S.; Avan, A.; Gholami, L.; Karimi, G. Auraptene Inhibits Migration and Invasion of Cervical and Ovarian Cancer Cells by Repression of Matrix Metalloproteinasas 2 and 9 Activity. J. Pharmacopunct. 2018, 21, 177–184.
- Hirose, Y.; Qiao, Z.; Murakami, A.; Ohigashi, H.; Tanaka, T.; Mori, H. Growth Inhibition of Colon Cancer Cells by Auraptene Is Not Correlated with the Modulation of Beta-Catenin-TCF Signaling. Cancer Res. 2004, 64, 318.
- Hayashi, K.; Suzuki, R.; Miyamoto, S.; Shin-Ichiroh, Y.; Kohno, H.; Sugie, S.; Takashima, S.; Tanaka, T. Citrus Auraptene Suppresses Azoxymethane-Induced Colonic Preneoplastic Lesions in C57BL/KsJ-db/dbMice. Nutr. Cancer 2007, 58, 75–84.
- Tanaka, T.; de Azevedo, M.B.M.; Durán, N.; Alderete, J.B.; Epifano, F.; Genovese, S.; Tanaka, M.; Tanaka, T.; Curini, M. Colorectal cancer chemoprevention by 2 β-cyclodextrin inclusion compounds of auraptene and 4′-geranyloxyferulic acid. Int. J. Cancer 2010, 126, 830–840.
- Krishnan, P.; Kleiner-Hancock, H. Effects of Auraptene on IGF-1 Stimulated Cell Cycle Progression in the Human Breast Cancer Cell Line, MCF-7. Int. J. Breast Cancer 2012, 2012, 502092.
- Krishnan, P.; Yan, K.J.; Windler, D.; Tubbs, J.; Grand, R.; Li, B.D.L.; Aldaz, C.M.; McLarty, J.; Kleiner-Hancock, H.E. Citrus auraptene suppresses cyclin D1 and significantly delays N-methyl nitrosourea induced mammary carcinogenesis in female Sprague-Dawley rats. BMC Cancer 2009, 9, 259.
- Lee, J.C.; Shin, E.A.; Kim, B.; Kim, B.-I.; Chitsazian-Yazdi, M.; Iranshahi, M.; Kim, S.-H. Auraptene Induces Apoptosis via Myeloid Cell Leukemia 1-Mediated Activation of Caspases in PC3 and DU145 Prostate Cancer Cells. Phytother. Res. 2017, 31, 891–898.
- Del Toro-Arreola, S.; Flores-Torales, E.; Torres-Lozano, C.; Del Toro-Arreola, A.; Tostado-Pelayo, K.; Ramirez-Dueñas, M.G.; Daneri-Navarro, A. Effect of d-limonene on immune response in BALB/c mice with lymphoma. Int. Immunopharmacol. 2005, 5, 829–838.
- Guo, X.-M.; Lu, Q.; Liu, Z.-J.; Wang, L.-F.; Feng, B.-A. Effects of D-limonene on leukemia cells HL-60 and K562 in vitro. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006, 14, 692–695.
- Yu, X.; Lin, H.; Wang, Y.; Lv, W.; Zhang, S.; Qian, Y.; Deng, X.; Feng, N.; Yu, H.; Qian, B. D-limonene exhibits antitumor activity by inducing autophagy and apoptosis in lung cancer. Onco Targets Ther. 2018, 11, 1833–1847.
- Zhang, X.-Z.; Wang, L.; Liu, D.-W.; Tang, G.-Y.; Zhang, H.-Y. Synergistic Inhibitory Effect of Berberine and d-Limonene on Human Gastric Carcinoma Cell Line MGC803. J. Med. Food 2014, 17, 955–962.
- Chaudhary, S.C.; Siddiqui, M.S.; Athar, M.; Alam, M.S. d-Limonene modulates inflammation, oxidative stress and Ras-ERK pathway to inhibit murine skin tumorigenesis. Hum. Exp. Toxicol. 2012, 31, 798–811.
- Jia, S.-S.; Xi, G.-P.; Zhang, M.; Chen, Y.-B.; Lei, B.; Dong, X.-S.; Yang, Y.-M. Induction of apoptosis by D-limonene is mediated by inactivation of Akt in LS174T human colon cancer cells. Oncol. Rep. 2012, 29, 349–354.
- Yeruva, L.; Pierre, K.J.; Elegbede, A.; Wang, R.C.; Carper, S.W. Perillyl alcohol and perillic acid induced cell cycle arrest and apoptosis in non small cell lung cancer cells. Cancer Lett. 2007, 257, 216–226.
- Bardon, S.; Foussard, V.; Fournel, S.; Loubat, A. Monoterpenes inhibit proliferation of human colon cancer cells by modulating cell cycle-related protein expression. Cancer Lett. 2002, 181, 187–194.
- Abbasi, R.; Efferth, T.; Kuhmann, C.; Opatz, T.; Hao, X.; Popanda, O.; Schmezer, P. The endoperoxide ascaridol shows strong differential cytotoxicity in nucleotide excision repair-deficient cells. Toxicol. Appl. Pharmacol. 2012, 259, 302–310.
- Bezerra, D.P.; Marinho-Filho, J.D.B.; Alves, A.P.N.N.; Pessoa, C.; De Moraes, M.O.; Pessoa, O.D.L.; Torres, M.C.M.; Silveira, E.R.; Viana, F.A.; Costa-Lotufo, L.V. Antitumor Activity of the Essential Oil from the Leaves ofCroton regelianusand Its Component Ascaridole. Chem. Biodivers. 2009, 6, 1224–1231.
- Fan, K.; Li, X.; Cao, Y.; Qi, H.; Li, L.; Zhang, Q.; Sun, H. Carvacrol inhibits proliferation and induces apoptosis in human colon cancer cells. Anti-Cancer Drugs 2015, 26, 813–823.
- Günes-Bayir, A.; Kiziltan, H.S.; Kocyigit, A.; Guler, E.M.; Karataş, E.; Toprak, A. Effects of natural phenolic compound carvacrol on the human gastric adenocarcinoma (AGS) cells in vitro. Anti-Cancer Drugs 2017, 28, 522–530.
- Al-Fatlawi, A.A.; Ahmad, A. Cytotoxicity and Pro-Apoptotic Activity of Carvacrol on Human Breast Cancer Cell Line MCF-7. World J. Pharm. Sci. 2014, 2, 1218–1223.
- Yin, Q.-H.; Yan, F.-X.; Zu, X.-Y.; Wu, Y.-H.; Wu, X.-P.; Liao, M.-C.; Deng, S.-W.; Yin, L.-L.; Zhuang, Y.-Z. Anti-proliferative and pro-apoptotic effect of carvacrol on human hepatocellular carcinoma cell line HepG-2. Cytotechnology 2012, 64, 43–51.
- Nalini, N.; Sivaranjani, A.; Sivagami, G. Chemopreventive effect of carvacrol on 1,2-dimethylhydrazine induced experimental colon carcinogenesis. J. Cancer Res. Ther. 2016, 12, 755–762.
- Koparal, A.T.; Zeytinoglu, M. Effects of Carvacrol on a Human Non-Small Cell Lung Cancer (NSCLC) Cell Line, A549. Anim. Cell Technol. Basic Appl. Asp. 2003, 13, 207–211.
- Patel, B.; Shah, V.R.; Bavadekar, S.A. Anti-proliferative effects of carvacrol on human prostate cancer cell line, LNCaP. FASEB J. 2012, 26, 1037.5.
- Arunasree, K.M. Anti-proliferative effects of carvacrol on a human metastatic breast cancer cell line, MDA-MB 231. Phytomedicine 2010, 17, 581–588.
- Baranauskaitė, J.; Kubiliene, A.; Marksa, M.; Petrikaite, V.; Vitkevicius, K.; Baranauskas, A.; Bernatoniene, J. The Influence of Different Oregano Species on the Antioxidant Activity Determined Using HPLC Postcolumn DPPH Method and Anticancer Activity of Carvacrol and Rosmarinic Acid. BioMed Res. Int. 2017, 2017, 1681392.
- Stammati, A.; Bonsi, P.; Zucco, F.; Moezelaar, R.; Alakomi, H.-L.; von Wright, A. Toxicity of Selected Plant Volatiles in Microbial and Mammalian Short-term Assays. Food Chem. Toxicol. 1999, 37, 813–823.
- Heidarian, E.; Keloushadi, M. Antiproliferative and Anti-invasion Effects of Carvacrol on PC3 Human Prostate Cancer Cells through Reducing pSTAT3, pAKT, and pERK1/2 Signaling Proteins. Int. J. Prev. Med. 2019, 10, 156.
- Khan, F.; Khan, I.; Farooqui, A.; Ansari, I.A. Carvacrol Induces Reactive Oxygen Species (ROS)-mediated Apoptosis Along with Cell Cycle Arrest at G0/G1 in Human Prostate Cancer Cells. Nutr. Cancer 2017, 69, 1075–1087.
- Khan, F.; Singh, V.K.; Saeed, M.; Kausar, M.A.; Ansari, I.A. Carvacrol Induced Program Cell Death and Cell Cycle Arrest in Androgen-Independent Human Prostate Cancer Cells via Inhibition of Notch Signaling. Anti-Cancer Agents Med. Chem. 2019, 19, 1588–1608.
- Zeytinoglu, H.; Incesu, Z.; Baser, K.H.C. Inhibition of DNA synthesis by Carvacrol in mouse myoblast cells bearing a human N-RAS oncogene. Phytomedicine 2003, 10, 292–299.
- Jayakumar, S.; Madankumar, A.; Asokkumar, S.; Raghunandhakumar, S.; Gokula Dhas, K.; Kamaraj, S.; Divya, M.G.J.; Devaki, T. Potential preventive effect of carvacrol against diethylnitrosamine-induced hepatocellular carcinoma in rats. Mol. Cell. Biochem. 2012, 360, 51–60.
- Jaafari, A.; Mouse, H.A.; Rakib, E.M.; M’Barek, L.A.; Tilaoui, M.; Benbakhta, C.; Boulli, A.; Abbad, A.; Zyad, A. Chemical composition and antitumor activity of different wild varieties of Moroccan thyme. Bras. J. Farm. 2007, 17, 477–491.
- Jaafari, A.; Mouse, H.A.; M’Bark, L.A.; Tilaoui, M.; Elhansali, M.; Lepoivre, M.; Aboufatima, R.; Melhaoui, A.; Chait, A.; Zyad, A. Differential Antitumor Effect of Essential Oils and Their Major Components of Thymus Broussonettii: Relationship to Cell Cycle and Apoptosis Induction. Herba Pol. J. 2009, 55, 36–50.
- Zeytinoglu, M.; Aydin, S.; Ozturk, Y.; Husnu, K.; Baser, C. Inhibitory Effects of Carvacrol on DMBA Induced Pulmonary Tumorigenesis in Rats. Acta Pharm. Turc. 1998, 40, 93–98.
- Rooney, S.; Ryan, M.F. Effects of alpha-hederin and thymoquinone, constituents of Nigella sativa, on human cancer cell lines. Anticancer Res. 2005, 25, 2199–2204.
- Bourgou, S.; Pichette, A.; Marzouk, B.; Legault, J. Bioactivities of black cumin essential oil and its main terpenes from Tunisia. S. Afr. J. Bot. 2010, 76, 210–216.
- Cecarini, V.; Quassinti, L.; Di Blasio, A.; Bonfili, L.; Bramucci, M.; Lupidi, G.; Cuccioloni, M.; Mozzicafreddo, M.; Angeletti, M.; Eleuteri, A.M. Effects of thymoquinone on isolated and cellular proteasomes. FEBS J. 2010, 277, 2128–2141.
- Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Targeting Nuclear Factor-κB Activation Pathway by Thymoquinone: Role in Suppression of Antiapoptotic Gene Products and Enhancement of Apoptosis. Mol. Cancer Res. 2008, 6, 1059–1070.
- Gali-Muhtasib, H.; Kuester, D.; Mawrin, C.; Bajbouj, K.; Diestel, A.; Ocker, M.; Habold, C.; Foltzer-Jourdainne, C.; Schoenfeld, P.; Peters, B.; et al. Thymoquinone Triggers Inactivation of the Stress Response Pathway Sensor CHEK1 and Contributes to Apoptosis in Colorectal Cancer Cells. Cancer Res. 2008, 68, 5609–5618.
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting Signal-Transducer-and-Activator-of-Transcription-3 for Prevention and Therapy of Cancer: Modern Target but Ancient So-lution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169.
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488.
- Li, F.; Rajendran, P.; Sethi, G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br. J. Pharmacol. 2010, 161, 541–554.
- Badary, O.A.; El-Din, A.M.G. Inhibitory effects of thymoquinone against 20-methylcholanthrene-induced fibrosarcoma tumorigenesis. Cancer Detect. Prev. 2001, 25, 362–368.
- Barron, J.; Benghuzzi, H.; Tucci, M. Effects of thymoquinone and selenium on the proliferation of mg 63 cells in tissue culture. Tech. Pap. ISA 2008, 44, 434–440.
- Roepke, M.; Diestel, A.; Bajbouj, K.; Walluscheck, D.; Schonfeld, P.; Roessner, A.; Schneider-Stock, R.; Gali-Muhtasib, H. Lack of p53 augments thymoquinone-induced apoptosis and caspase activation in human osteosarcoma cells. Cancer Biol. Ther. 2007, 6, 160–169.
- Peng, L.; Liu, A.; Shen, Y.; Xu, H.-Z.; Yang, S.-Z.; Ying, X.-Z.; Liao, W.; Liu, H.-X.; Lin, Z.-Q.; Chen, Q.-Y.; et al. Antitumor and anti-angiogenesis effects of thymoquinone on osteosarcoma through the NF-κB pathway. Oncol. Rep. 2012, 29, 571–578.
- Yazan, L.S.; Ng, W.K.; Al-naqeeb, G.; Ismail, M. Cytotoxicity of Thymoquinone (TQ) from Nigella Sativa Towards Human Cervical Carcinoma Cells (HeLa). J. Pharm. Res. 2009, 2, 585–589.
- El-Najjar, N.; Chatila, M.; Moukadem, H.; Vuorela, H.; Ocker, M.; Gandesiri, M.; Schneider-Stock, R.; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2009, 15, 183–195.
- Zubair, H.; Khan, H.Y.; Sohail, A.; Azim, S.; Ullah, M.F.; Ahmad, A.; Sarkar, F.H.; Hadi, S.M. Redox cycling of endogenous copper by thymoquinone leads to ROS-mediated DNA breakage and consequent cell death: Putative anticancer mechanism of antioxidants. Cell Death Dis. 2013, 4, e660.
- Talib, W.H.; Abukhader, M.M. Combinatorial Effects of Thymoquinone on the Anticancer Activity and Hepatotoxicity of the Prodrug CB 1954. Sci. Pharm. 2013, 81, 519–530.
- Richards, L.R.; Jones, P.; Hughes, J.; Benghuzzi, H.; Tucci, M. LNCaP cells exposed to ceramic drug delivery treatment with epigallocatechin-3-gallate, thymoquinone, and tannic acid. Biomed. Sci. Instrum. 2007, 43, 242–247.
- Richards, L.R.; Jones, P.; Benghuzzi, H.; Tucci, M. A Comparison of the Morphological Changes Associated with Conventional and Sustained Treatment with Epigallocatechin-3-Gallate, Thymoquinone, and Tannic Acid on LNCAP Cells. Tech. Pap. ISA 2008, 44, 465–470.
- Koka, P.S.; Mondal, D.; Schultz, M.; Abdel-Mageed, A.B.; Agrawal, K.C. Studies on molecular mechanisms of growth inhibitory effects of thymoquinone against prostate cancer cells: Role of reactive oxygen species. Exp. Biol. Med. 2010, 235, 751–760.
- El-Mahdy, M.A.; Zhu, Q.; Wang, Q.-E.; Wani, G.; Wani, A.A. Thymoquinone induces apoptosis through activation of caspase-8 and mitochondrial events in p53-null myeloblastic leukemia HL-60 cells. Int. J. Cancer 2005, 117, 409–417.
- Hassan, S.A.; Ahmed, W.A.; Galeb, F.M.; El-Taweel, M.A.; Abu-Bedair, F.A. In Vitro Challenge using Thymoquinone on Hepatocellular Carcinoma (HepG2) Cell Line. Iran. J. Pharm. Res 2008, 7, 283–290.
- Gali-Muhtasib, H.U.; Abou-Kheir, W.G.; Kheir, L.A.; Darwiche, N.; Crooks, P.A. Molecular pathway for thymoquinone-induced cell-cycle arrest and apoptosis in neoplastic keratinocytes. Anti-Cancer Drugs 2004, 15, 389–399.
- Rajput, S.; Kumar, B.N.P.; Dey, K.K.; Pal, I.; Parekh, A.; Mandal, M. Molecular targeting of Akt by thymoquinone promotes G1 arrest through translation inhibition of cyclin D1 and induces apoptosis in breast cancer cells. Life Sci. 2013, 93, 783–790.
- Gurung, R.L.; Ni Lim, S.; Khaw, A.K.; Soon, J.F.F.; Shenoy, K.; Ali, S.M.; Jayapal, M.; Sethu, S.; Baskar, R.; Hande, M.P. Thymoquinone Induces Telomere Shortening, DNA Damage and Apoptosis in Human Glioblastoma Cells. PLoS ONE 2010, 5, e12124.
- Paramasivam, A.; Sambantham, S.; Shabnam, J.; Raghunandhakumar, S.; Anandan, B.; Rajiv, R.; Priyadharsini, J.V.; Jayaraman, G. Anti-cancer effects of thymoquinone in mouse neuroblastoma (Neuro-2a) cells through caspase-3 activation with down-regulation of XIAP. Toxicol. Lett. 2012, 213, 151–159.
- Torres, M.P.; Ponnusamy, M.P.; Chakraborty, S.; Smith, L.M.; Das, S.; Arafat, H.A.; Batra, S.K. Effects of Thymoquinone in the Expression of Mucin 4 in Pancreatic Cancer Cells: Implications for the Development of Novel Cancer Therapies. Mol. Cancer Ther. 2010, 9, 1419–1431.
- Al-Shabanah, O.A.; Badary, O.A.; Nagi, M.N.; Al-Gharably, N.M.; Al-Rikabi, A.C.; Al-Bekairi, A.M. Thymoquinone protects against doxorubicin-induced cardiotoxicity without compromising its antitumor activity. J. Exp. Clin. Cancer Res. 1998, 17, 193–198.
- Nagi, M.N.; Almakki, H.A. Thymoquinone supplementation induces quinone reductase and glutathione transferase in mice liver: Possible role in protection against chemical carcinogenesis and toxicity. Phytother. Res. 2009, 23, 1295–1298.
- Salem, M.L.; Alenzi, F.Q.; Attia, W.Y. Thymoquinone, the active ingredient ofNigella sativaseeds, enhances survival and activity of antigen-specific CD8-positive T cellsin vitro. Br. J. Biomed. Sci. 2011, 68, 131–137.
- Attoub, S.; Sperandio, O.; Raza, H.; Arafat, K.; Al-Salam, S.; Al Sultan, M.A.; Al Safi, M.; Takahashi, T.; Adem, A. Thymoquinone as an anticancer agent: Evidence from inhibition of cancer cells viability and invasion in vitro and tumor growth in vivo. Fundam. Clin. Pharmacol. 2013, 27, 557–569.
- Lang, M.; Borgmann, M.; Oberhuber, G.; Evstatiev, R.; Jimenez, K.; Dammann, K.W.; Jambrich, M.; Khare, V.; Campregher, C.; Ristl, R.; et al. Thymoquinone attenuates tumor growth in ApcMin mice by interference with Wnt-signaling. Mol. Cancer 2013, 12, 1–13.
- AbdElfadil, E.; Cheng, Y.-H.; Bau, D.-T.; Ting, W.-J.; Chen, L.-M.; Hsu, H.-H.; Lin, Y.-M.; Chen, R.-J.; Tsai, F.-J.; Tsai, C.-H.; et al. Thymoquinone Induces Apoptosis in Oral Cancer Cells Through P38β Inhibition. Am. J. Chin. Med. 2013, 41, 683–696.
- Das, S.; Dey, K.K.; Dey, G.; Pal, I.; Majumder, A.; MaitiChoudhury, S.; Kundu, S.C.; Mandal, M. Antineoplastic and Apoptotic Potential of Traditional Medicines Thymoquinone and Diosgenin in Squamous Cell Carcinoma. PLoS ONE 2012, 7, e46641.
- Alhosin, M.; Ibrahim, A.; Boukhari, A.; Sharif, T.; Gies, J.-P.; Auger, C.; Schini-Kerth, V.B. Anti-neoplastic agent thymoquinone induces degradation of α and β tubulin proteins in human cancer cells without affecting their level in normal human fibroblasts. Investig. New Drugs 2011, 30, 1813–1819.
- Kus, G.; Ozkurt, M.; Kabadere, S.; Erkasap, N.; Goger, G.; Demirci, F. Antiproliferative and antiapoptotic effect of thymoquinone on cancer cells in vitro. Bratisl. Med. J. 2018, 119, 312–316.
- Liou, Y.F.; Chen, P.N.; Chu, S.C.; Kao, S.H.; Chang, Y.Z.; Hsieh, Y.S.; Chang, H.R. Thymoquinone suppresses the proliferation of renal cell carcinoma cells via reactive oxygen species-induced apoptosis and reduces cell stemness. Environ. Toxicol. 2019, 34, 1208–1220.
- Zhang, Y.; Fan, Y.; Huang, S.; Wang, G.; Han, R.; Lei, F.; Luo, A.; Jing, X.; Zhao, L.; Gu, S.; et al. Thymoquinone inhibits the metastasis of renal cell cancer cells by inducing autophagy via AMPK/mTOR signaling pathway. Cancer Sci. 2018, 109, 3865–3873.
- Sotillo, W.S.; Villagomez, R.; Smiljanic, S.; Huang, X.; Malakpour, A.; Kempengren, S.; Rodrigo, G.; Almanza, G.; Sterner, O.; Oredsson, S. Anti-cancer stem cell activity of a sesquiterpene lactone isolated from Ambrosia arborescens and of a synthetic derivative. PLoS ONE 2017, 12, e0184304.
- Hehner, S.P.; Heinrich, M.; Bork, P.M.; Vogt, M.; Ratter, F.; Lehmann, V.; Schulze-Osthoff, K.; Dröge, W.; Schmitz, M.L. Sesquiterpene Lactones Specifically Inhibit Activation of NF-κB by Preventing the Degradation of IκB-α and IκB-β. J. Biol. Chem. 1998, 273, 1288–1297.
- Shoaib, M.; Shah, I.; Ali, N.; Adhikari, A.; Tahir, M.N.; Shah, S.W.A.; Ishtiaq, S.; Khan, J.; Khan, S.; Umer, M.N. Sesquiterpene lactone! a promising antioxidant, anticancer and moderate antinociceptive agent from Artemisia macrocephala jacquem. BMC Complement. Altern. Med. 2017, 17, 27.
- Gach, K.; Długosz, A.; Janecka, A. The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn. Schmiedeberg’s. Arch. Pharmacol. 2015, 388, 477–486.
- Mathema, V.B.; Koh, Y.-S.; Thakuri, B.C.; Sillanpää, M. Parthenolide, a Sesquiterpene Lactone, Expresses Multiple Anti-cancer and Anti-inflammatory Activities. Inflammation 2012, 35, 560–565.
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678.
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to trail by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell. Physiol. 2011, 226, 1632–1641.
- Liu, M.; Xiao, C.; Sun, M.; Tan, M.; Hu, L.; Yu, Q. Parthenolide Inhibits STAT3 Signaling by Covalently Targeting Janus Kinases. Molecules 2018, 23, 1478.
- Wen, J.; You, K.-R.; Lee, S.-Y.; Song, C.-H.; Kim, D.-G. Oxidative Stress-mediated Apoptosis: The Anticancer Effect of the Ses-quiterpene Lactone Parthenolide. J. Biol. Chem. 2002, 277, 38954–38964.
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955.
- Hsu, Y.-L.; Wu, L.-Y.; Kuo, P.-L. Dehydrocostuslactone, a Medicinal Plant-Derived Sesquiterpene Lactone, Induces Apoptosis Coupled to Endoplasmic Reticulum Stress in Liver Cancer Cells. J. Pharmacol. Exp. Ther. 2009, 329, 808–819.
- Huang, P.-R.; Yeh, Y.-M.; Wang, T.-C.V. Potent inhibition of human telomerase by helenalin. Cancer Lett. 2005, 227, 169–174.
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol. Immunol. 2009, 46, 2892–2901.
- Lyß, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The Anti-inflammatory Sesquiterpene Lactone Helenalin Inhibits the Transcription Factor NF-κB by Directly Targeting P65. J. Biol. Chem. 1998, 273, 33508–33516.
- Lim, C.B.; Fu, P.Y.; Ky, N.; Zhu, H.S.; Feng, X.L.; Li, J.; Srinivasan, K.G.; Hamza, M.S.; Zhao, Y. NF-κB p65 repression by the sesquiterpene lactone, Helenalin, contributes to the induction of autophagy cell death. BMC Complement. Altern. Med. 2012, 12, 93.
- Li, H.; Li, M.; Wang, G.; Shao, F.; Chen, W.; Xia, C.; Wang, S.; Li, Y.; Zhou, G.; Liu, Z. EM23, A Natural Sesquiterpene Lactone from Elephantopus mollis, Induces Apoptosis in Human Myeloid Leukemia Cells through Thioredoxin- and Reactive Oxygen Species-Mediated Signaling Pathways. Front. Pharmacol. 2016, 7, 77.
- Ivanescu, B.; Miron, A.; Corciova, A. Sesquiterpene Lactones fromArtemisiaGenus: Biological Activities and Methods of Analysis. J. Anal. Methods Chem. 2015, 2015, 247685.
- Wang, Q.; Wu, L.-M.; Zhao, Y.; Zhang, X.-L.; Wang, N.-P. The anticancer effect of artesunate and its mechanism. Yao Xue Xue Bao 2002, 37, 477–478.
- Jiang, W.; Huang, Y.; Wang, J.-P.; Yu, X.-Y.; Zhang, L.-Y. The Synergistic Anticancer Effect of Artesunate Combined with Allicin in Osteosarcoma Cell Line in Vitro and in Vivo. Asian Pac. J. Cancer Prev. 2013, 14, 4615–4619.
- Roh, J.-L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262.
- Zheng, L.; Pan, J. The Anti-malarial Drug Artesunate Blocks Wnt/β-catenin Pathway and Inhibits Growth, Migration and Invasion of Uveal Melanoma Cells. Curr. Cancer Drug Targets 2018, 18, 988–998.
- Verma, S.; Das, P.; Kumar, V.L. Chemoprevention by artesunate in a preclinical model of colorectal cancer involves down regulation of β-catenin, suppression of angiogenesis, cellular proliferation and induction of apoptosis. Chem. Biol. Interact. 2017, 278, 84–91.
- Jiang, F.; Zhou, J.Y.; Zhang, D.; Liu, M.H.; Chen, Y.G. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunate-induced apoptosis. Int. J. Mol. Med. 2018, 42, 1295–1304.
- Chen, X.; Wong, Y.K.; Lim, T.K.; Lim, W.H.; Lin, Q.; Wang, J.; Hua, Z. Artesunate Activates the Intrinsic Apoptosis of HCT116 Cells through the Suppression of Fatty Acid Synthesis and the NF-κB Pathway. Molecules 2017, 22, 1272.
- Li, L.-N.; Zhang, H.-D.; Yuan, S.-J.; Tian, Z.-Y.; Wang, L.; Sun, Z.-X. Artesunate attenuates the growth of human colorectal carcinoma and inhibits hyperactive Wnt/β-catenin pathway. Int. J. Cancer 2007, 121, 1360–1365.
- Ji, P.; Huang, H.; Yuan, S.; Wang, L.; Wang, S.; Chen, Y.; Feng, N.; Veroniaina, H.; Wu, Z.; Wu, Z.; et al. ROS-Mediated Apoptosis and Anticancer Effect Achieved by Artesunate and Auxiliary Fe(II) Released from Ferriferous Oxide-Containing Recombinant Apoferritin. Adv. Healthcare Mater. 2019, 8, e1900911.
- Greenshields, A.L.; Fernando, W.; Hoskin, D.W. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp. Mol. Pathol. 2019, 107, 10–22.
- Wang, X.; Du, Q.; Mao, Z.; Fan, X.; Hu, B.; Wang, Z.; Chen, Z.; Jiang, X.; Wang, Z.; Lei, N.; et al. Combined treatment with artesunate and bromocriptine has synergistic anticancer effects in pituitary adenoma cell lines. Oncotarget 2017, 8, 45874–45887.
- Wang, Z.; Wang, Q.; He, T.; Li, W.; Liu, Y.; Fan, Y.; Wang, Y.; Wang, Q.; Chen, J. The combination of artesunate and carboplatin exerts a synergistic anti-tumour effect on non-small cell lung cancer. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1083–1091.
- Ma, H.; Yao, Q.; Zhang, A.-M.; Lin, S.; Wang, X.; Wu, L.; Sun, J.-G.; Chen, Z.-T. The Effects of Artesunate on the Expression of EGFR and ABCG2 in A549 Human Lung Cancer Cells and a Xenograft Model. Molecules 2011, 16, 10556–10569.
- Beccafico, S.; Morozzi, G.; Marchetti, M.C.; Riccardi, C.; Sidoni, A.; Donato, R.; Sorci, G. Artesunate induces ROS- and p38 MAPK-mediated apoptosis and counteracts tumor growthin vivoin embryonal rhabdomyosarcoma cells. Carcinogenesis 2015, 36, 1071–1083.
- Yang, Y.; Wu, N.; Wu, Y.; Chen, H.; Qiu, J.; Qian, X.; Zeng, J.; Chiu, K.; Gao, Q.; Zhuang, J. Artesunate induces mitochondria-mediated apoptosis of human retinoblastoma cells by upregulating Kruppel-like factor. Cell Death Dis. 2019, 10, 1–12.
- Liu, Y.; Cui, Y. Synergism of Cytotoxicity Effects of Triptolide and Artesunate Combination Treatment in Pancreatic Cancer Cell Lines. Asian Pac. J. Cancer Prev. 2013, 14, 5243–5248.
- Yao, X.; Zhao, C.-R.; Yin, H.; Wang, K.; Gao, J.-J. Synergistic antitumor activity of sorafenib and artesunate in hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2020, 41, 1609–1620.
- Ilamathi, M.; Santhosh, S.; Sivaramakrishnan, V. Artesunate as an Anti-Cancer Agent Targets Stat-3 and Favorably Suppresses Hepatocellular Carcinoma. Curr. Top. Med. Chem. 2016, 16, 2453–2463.
- Zhang, L.; Qian, H.; Sha, M.; Luan, Z.; Lin, M.; Yuan, D.; Li, X.; Huang, J.; Ye, L. Downregulation of HOTAIR Expression Mediated Anti-Metastatic Effect of Artesunate on Cervical Cancer by Inhibiting COX-2 Expression. PLoS ONE 2016, 11, e0164838.
- Liu, L.; Zuo, L.-F.; Zuo, J.; Wang, J. Artesunate induces apoptosis and inhibits growth of Eca109 and Ec9706 human esophageal cancer cell lines in vitro and in vivo. Mol. Med. Rep. 2012, 12, 1465–1472.
- Kim, C.; Lee, J.H.; Kim, S.-H.; Sethi, G.; Ahn, K.S. Artesunate suppresses tumor growth and induces apoptosis through the modulation of multiple oncogenic cascades in a chronic myeloid leukemia xenograft mouse model. Oncotarget 2015, 6, 4020–4035.
- Zhou, X.; Sun, W.-J.; Wang, W.-M.; Chen, K.; Zheng, J.-H.; Lu, M.-D.; Li, P.-H.; Zheng, Z.-Q. Artesunate inhibits the growth of gastric cancer cells through the mechanism of promoting oncosis both in vitro and in vivo. Anti-Cancer Drugs 2013, 24, 920–927.
- Jiang, Z.; Chai, J.; Chuang, H.H.F.; Li, S.; Wang, T.; Cheng, Y.; Chen, W.; Zhou, D. Artesunate induces G0/G1 cell cycle arrest and iron-mediated mitochondrial apoptosis in A431 human epidermoid carcinoma cells. Anti-Cancer Drugs 2012, 23, 606–613.
- Ilamathi, M.; Prabu, P.C.; Ayyappa, K.A.; Sivaramakrishnan, V. Artesunate obliterates experimental hepatocellular carcinoma in rats through suppression of IL-6-JAK-STAT signalling. Biomed. Pharmacother. 2016, 82, 72–79.
- Li, Y.; Feng, L.; Jiang, W.; Shan, N.; Wang, X. Artesunate possesses anti-leukemia properties that can be enhanced by arsenic trioxide. Leuk. Lymphoma 2013, 55, 1366–1372.
- Jiang, Z.; Jacob, J.A.; Loganathachetti, D.S.; Nainangu, P.; Chen, B. β-Elemene: Mechanistic Studies on Cancer Cell Interaction and Its Chemosensitization Effect. Front. Pharmacol. 2017, 8, 105.
- Wang, G.; Li, X.; Huang, F.; Zhao, J.; Ding, H.; Cunningham, C.; Coad, J.E.; Flynn, D.C.; Reed, E.; Li, Q.Q. Antitumor effect of β-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell. Mol. Life Sci. 2005, 62, 881–893.
- Tamgue, O.; Lei, M. Triptolide promotes senescence of prostate cancer cells through induction of histone methylation and heterochromatin formation. Asian Pac. J. Cancer Prev. 2017, 18, 2519–2526.
- Huang, W.; He, T.; Chai, C.; Yang, Y.; Zheng, Y.; Zhou, P.; Qiao, X.; Zhang, B.; Liu, Z.; Wang, J.; et al. Triptolide Inhibits the Proliferation of Prostate Cancer Cells and Down-Regulates SUMO-Specific Protease 1 Expression. PLoS ONE 2012, 7, e37693.
- Titov, D.V.; Gilman, B.; He, Q.-L.; Bhat, S.; Low, W.-K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188.
- Pan, J. RNA polymerase—An important molecular target of triptolide in cancer cells. Cancer Lett. 2010, 292, 149–152.
- Manzo, S.G.; Zhou, Z.-L.; Wang, Y.-Q.; Marinello, J.; He, J.X.; Li, Y.-C.; Ding, J.; Capranico, G.; Miao, Z.-H. Natural Product Triptolide Mediates Cancer Cell Death by Triggering CDK7-Dependent Degradation of RNA Polymerase II. Cancer Res. 2012, 72, 5363–5373.
- Chen, Y.-W.; Lin, G.-J.; Chuang, Y.-P.; Chia, W.-T.; Hueng, D.-Y.; Lin, C.-K.; Nieh, S.; Sytwu, H.-K. Triptolide circumvents drug-resistant effect and enhances 5-fluorouracil antitumor effect on KB cells. Anti-Cancer Drugs 2010, 21, 502–513.
- Yi, J.-M.; Huan, X.-J.; Song, S.-S.; Zhou, H.; Wang, Y.-Q.; Miao, Z.-H. Triptolide Induces Cell Killing in Multidrug-Resistant Tumor Cells via CDK7/RPB1 Rather than XPB or P44. Mol. Cancer Ther. 2016, 15, 1495–1503.
- Reno, T.A.; Kim, J.Y.; Raz, D.J. Triptolide Inhibits Lung Cancer Cell Migration, Invasion, and Metastasis. Ann. Thorac. Surg. 2015, 100, 1817–1825.
- Hamdi, A.M.; Jiang, Z.-Z.; Guerram, M.; Yousef, B.A.; Hassan, H.M.; Ling, J.-W.; Zhang, L.-Y. Biochemical and computational evaluation of Triptolide-induced cytotoxicity against NSCLC. Biomed. Pharmacother. 2018, 103, 1557–1566.
- Jiang, W.; Chen, M.; Xiao, C.; Yang, W.; Qin, Q.; Tan, Q.; Liang, Z.; Liao, X.; Mao, A.; Wei, C. Triptolide Suppresses Growth of Breast Cancer by Targeting HMGB1 in Vitro and in Vivo. Biol. Pharm. Bull. 2019, 42, 892–899.
- Xiong, J.; Su, T.; Qu, Z.; Yang, Q.; Wang, Y.; Li, J.; Zhou, S. Triptolide has anticancer and chemosensitization effects by down-regulating Akt activation through the MDM2/REST pathway in human breast cancer. Oncotarget 2016, 7, 23933–23946.
- Qin, G.; Li, P.; Xue, Z. Triptolide induces protective autophagy and apoptosis in human cervical cancer cells by downregulating Akt/mTOR activation. Oncol. Lett. 2018, 16, 3929–3934.
- Varghese, E.; Samuel, S.M.; Varghese, S.; Cheema, S.; Mamtani, R.; Büsselberg, D. Triptolide Decreases Cell Proliferation and Induces Cell Death in Triple Negative MDA-MB-231 Breast Cancer Cells. Biomolecules 2018, 8, 163.
- Hu, H.; Zhu, S.; Tong, Y.; Huang, G.; Tan, B.; Yang, L. Antitumor activity of triptolide in SKOV3 cells and SKOV3/DDP in vivo and in vitro. Anti-Cancer Drugs 2020, 31, 483–491.
- Hu, H.; Huang, G.; Wang, H.; Li, X.; Wang, X.; Feng, Y.; Tan, B.; Chen, T. Inhibition effect of triptolide on human epithelial ovarian cancer via adjusting cellular immunity and angiogenesis. Oncol. Rep. 2017, 39, 1191–1196.
- Li, S.; Qu, Y.; Shen, X.-Y.; Ouyang, T.; Fu, W.-B.; Luo, T.; Wang, H.-Q. Multiple Signal Pathways Involved in Crocetin-Induced Apoptosis in KYSE-150 Cells. Pharmacology 2019, 103, 263–272.
- Li, S.; Shen, X.-Y.; Ouyang, T.; Qu, Y.; Luo, T.; Wang, H.-Q. Synergistic anticancer effect of combined crocetin and cisplatin on KYSE-150 cells via p53/p21 pathway. Cancer Cell Int. 2017, 17, 1–11.
- Ling-Ping, K.; Jiang, S.; Jiang, W.; Zhou, Y.; Shen, X.-Y.; Luo, T.; Kong, L.-P.; Wang, H.-Q. Anticancer effects of crocetin in human esophageal squamous cell carcinoma KYSE-150 cells. Oncol. Lett. 2015, 9, 1254–1260.
- Moradzadeh, M.; Ghorbani, A.; Erfanian, S.; Mohaddes, S.T.; Rahimi, H.; Karimiani, E.G.; Mashkani, B.; Chiang, S.; El-Khamisy, S.F.; Tabarraei, A.; et al. Study of the mechanisms of crocetin-induced differentiation and apoptosis in human acute promyelocytic leukemia cells. J. Cell. Biochem. 2019, 120, 1943–1957.
- Sajjadi, M.; Bathaie, Z. Comparative Study on The Preventive Effect of Saffron Carotenoids, Crocin and Crocetin, in NMU-Induced Breast Cancer in Rats. Cell J. 2016, 19, 94–101.
- Bathaie, S.Z.; Hoshyar, R.; Miri, H.; Sadeghizadeh, M. Anticancer effects of crocetin in both human adenocarcinoma gastric cancer cells and rat model of gastric cancer. Biochem. Cell Biol. 2013, 91, 397–403.
- Bathaie, S.Z.; Bolhassani, A.; Hoshyar, R.; Ranjbar, B.; Sabouni, F.; Moosavi-Movahedi, A.-A. Interaction of Saffron Carotenoids as Anticancer Compounds with ctDNA, Oligo (dG.dC)15, and Oligo (dA.dT)15. DNA Cell Biol. 2007, 26, 533–540.
- Kanakis, C.; Tarantilis, P.A.; Tajmir-Riahi, H.-A.; Polissiou, M.G. Interaction of tRNA with Safranal, Crocetin, and Dimethylcrocetin. J. Biomol. Struct. Dyn. 2007, 24, 537–545.
- Magesh, V.; DurgaBhavani, K.; Senthilnathan, P.; Rajendran, P.; Sakthisekaran, D. In vivoprotective effect of crocetin on benzo(a)pyrene-induced lung cancer in Swiss albino mice. Phytother. Res. 2009, 23, 533–539.
- Magesh, V.; Singh, J.P.V.; Selvendiran, K.; Ekambaram, G.; Sakthisekaran, D. Antitumour activity of crocetin in accordance to tumor incidence, antioxidant status, drug metabolizing enzymes and histopathological studies. Mol. Cell. Biochem. 2006, 287, 127–135.
- Zhong, Y.-J.; Shi, F.; Zheng, X.-L.; Wang, Q.; Yang, L.; Sun, H.; He, F.; Zhang, L.; Lin, Y.; Qin, Y.; et al. Crocetin induces cytotoxicity and enhances vincristine-induced cancer cell death via p53-dependent and -independent mechanisms. Acta Pharmacol. Sin. 2011, 32, 1529–1536.
- Gutheil, W.; Reed, G.; Ray, A.; Anant, S.; Dhar, A. Crocetin: An Agent Derived from Saffron for Prevention and Therapy for Cancer. Curr. Pharm. Biotechnol. 2012, 13, 173–179.
- Sakthivel, R.; Malar, D.S.; Devi, K.P. Phytol shows anti-angiogenic activity and induces apoptosis in A549 cells by depolarizing the mitochondrial membrane potential. Biomed. Pharmacother. 2018, 105, 742–752.
- Song, Y.; Cho, S.K. Phytol Induces Apoptosis and ROS-Mediated Protective Autophagy in Human Gastric Adenocarcinoma AGS Cells. Biochem. Anal. Biochem. 2015, 4, 1.
- Pejin, B.; Kojić, V.; Bogdanovic, G. An insight into the cytotoxic activity of phytol atin vitroconditions. Nat. Prod. Res. 2014, 28, 2053–2056.
- Sheeja, L.; Lakshmi, D.; Bharadwaj, S.; Parveen, K.S. Anticancer Activity of Phytol Purified from Gracilaria Edulis against Human Breast Cancer Cell Line (MCF-7). Int. J. Curr. Sci. 2016, 19, 36–46.
- Duval, R.E.; Harmand, P.-O.; Jayat-Vignoles, C.; Cook-Moreau, J.; Pinon, A.; Delage, C.; Simon, A. Differential involvement of mitochondria during ursolic acid-induced apoptotic process in HaCaT and M4Beu cells. Oncol. Rep. 2008, 19, 145–149.
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Ursolic acid in cancer prevention and treatment: Molecular targets, pharmacokinetics and clinical studies. Biochem. Pharmacol. 2013, 85, 1579–1587.
- Tang, X.; Gao, J.; Chen, J.; Fang, F.; Wang, Y.; Dou, H.; Xu, Q.; Qian, Z. Inhibition of ursolic acid on calcium-induced mitochondrial permeability transition and release of two proapoptotic proteins. Biochem. Biophys. Res. Commun. 2005, 337, 320–324.
- Shyu, M.-H.; Kao, T.-C.; Yen, G.-C. Oleanolic Acid and Ursolic Acid Induce Apoptosis in HuH7 Human Hepatocellular Carcinoma Cells through a Mitochondrial-Dependent Pathway and Downregulation of XIAP. J. Agric. Food Chem. 2010, 58, 6110–6118.
- Saraswati, S.; Agrawal, S.S.; Alhaider, A.A. Ursolic acid inhibits tumor angiogenesis and induces apoptosis through mitochondrial-dependent pathway in Ehrlich ascites carcinoma tumor. Chem. Biol. Interact. 2013, 206, 153–165.
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deręgowska, A.; Wnuk, M. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis 2017, 22, 800–815.
- Tu, H.-Y.; Huang, A.-M.; Wei, B.-L.; Gan, K.-H.; Hour, T.-C.; Yang, S.-C.; Pu, Y.-S.; Lin, C.-N. Ursolic acid derivatives induce cell cycle arrest and apoptosis in NTUB1 cells associated with reactive oxygen species. Bioorganic Med. Chem. 2009, 17, 7265–7274.
- Wu, C.-C.; Huang, Y.-F.; Hsieh, C.-P.; Chueh, P.-J.; Chen, Y.-L. Combined Use of Zoledronic Acid Augments Ursolic Acid-Induced Apoptosis in Human Osteosarcoma Cells through Enhanced Oxidative Stress and Autophagy. Molecules 2016, 21, 1640.
- Kim, K.H.; Seo, H.S.; Choi, H.S.; Choi, I.H.; Shin, Y.C.; Ko, S.-G. Induction of apoptotic cell death by ursolic acid through mitochondrial death pathway and extrinsic death receptor pathway in MDA-MB-231 cells. Arch. Pharmacal Res. 2011, 34, 1363–1372.
- Nam, H.; Kim, M.-M. Ursolic acid induces apoptosis of SW480 cells via p53 activation. Food Chem. Toxicol. 2013, 62, 579–583.
- Manu, K.A.; Kuttan, G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-κB mediated activation of bcl-2 in B16F-10 melanoma cells. Int. Immunopharmacol. 2008, 8, 974–981.
- Yu, Y.-X.; Gu, Z.-L.; Yin, J.-L.; Chou, W.-H.; Kwok, C.-Y.; Qin, Z.-H.; Liang, Z.-Q. Ursolic acid induces human hepatoma cell line SMMC-7721 apoptosis via p53-dependent pathway. Chin. Med. J. 2010, 123, 1915–1923.
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov. Today 2009, 14, 885–890.
- Fulda, S.; Scaffidi, G.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.-M. Activation of Mitochondria and Release of Mitochondrial Apoptogenic Factors by Betulinic Acid. J. Biol. Chem. 1998, 273, 33942–33948.
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464.
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88.
- Tan, Y.M.; Yu, R.; Pezzuto, J.M. Betulinic Acid-Induced Programmed Cell Death in Human Melanoma Cells Involves Mito-gen-Activated Protein Kinase Activation. Clin. Cancer Res. 2003, 9, 2866–2875.
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as Potentially Cytotoxic Compounds. Molecules 2015, 20, 1610–1625.
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Orfanos, C.E.; Geilen, C.C.; Wieder, T.; Sturm, I.; Daniel, P.T. The Bax/Bcl-2 Ratio Determines the Susceptibility of Human Melanoma Cells to CD95/Fas-Mediated Apoptosis. J. Investig. Dermatol. 2001, 117, 333–340.
- Rieber, M.; Rieber, M.S. Induction of p53Without Increase in p21WAF1 in Betulinic Acid-Mediated Cell Death Is Preferential for Human Metastatic Melanoma. DNA Cell Biol. 1998, 17, 399–406.
- Li, Y.; He, K.; Huang, Y.; Zheng, D.; Gao, C.; Cui, L.; Jin, Y.-H. Betulin induces mitochondrial cytochrome c release associated apoptosis in human cancer cells. Mol. Carcinog. 2010, 49, 630–640.
- Wang, D.Y.; Liu, J.; Yin, M.Z.; Li, X.T.; Lou, G.; Liu, Y.D.; Chen, X.W. Betulin Induces Apoptosis of HeLa Cell Lines in Vitro and Its Possible Mechanism. Tumor 2012, 32, 234–238.
- Zhao, J.; Li, R.; Pawlak, A.; Henklewska, M.; Sysak, A.; Wen, L.; Yi, J.-E.; Obmińska-Mrukowicz, B. Antitumor Activity of Betulinic Acid and Betulin in Canine Cancer Cell Lines. In Vivo 2018, 32, 1081–1088.
- Xu, T.; Pang, Q.; Wang, Y.; Yan, X. Betulinic acid induces apoptosis by regulating PI3K/Akt signaling and mitochondrial pathways in human cervical cancer cells. Int. J. Mol. Med. 2017, 40, 1669–1678.
- Luo, R.; Fang, D.; Chu, P.; Wu, H.; Zhang, Z.; Tang, Z. Multiple molecular targets in breast cancer therapy by betulinic acid. Biomed. Pharmacother. 2016, 84, 1321–1330.
- Foo, J.B.; Yazan, L.S.; Tor, Y.S.; Wibowo, A.; Ismail, N.; How, C.W.; Armania, N.; Loh, S.P.; Ismail, I.S.; Cheah, Y.K.; et al. Induction of cell cycle arrest and apoptosis by betulinic acid-rich fraction from Dillenia suffruticosa root in MCF-7 cells involved p53/p21 and mitochondrial signalling pathway. J. Ethnopharmacol. 2015, 166, 270–278.
- Jiang, W.; Li, X.; Dong, S.; Zhou, W. Betulinic acid in the treatment of tumour diseases: Application and research progress. Biomed. Pharmacother. 2021, 142, 111990.
- Zhan, X.K.; Li, J.L.; Zhang, S.; Xing, P.Y.; Xia, M.F. Betulinic acid exerts potent antitumor effects on paclitaxel-resistant human lung carcinoma cells (H460) via G2/M phase cell cycle arrest and induction of mitochondrial apoptosis. Oncol. Lett. 2018, 16, 3628–3634.
- Pitchai, D.; Roy, A.; Ignatius, C. In vitro evaluation of anticancer potentials of lupeol isolated from Elephantopus scaber L. on MCF-7 cell line. J. Adv. Pharm. Technol. Res. 2014, 5, 179–184.
- Babu, S.T. Study on the Anti Metastatic and Anticancer Activity of Triterpene Compound Lupeol in Human Lung Cancer. Int. J. Pharm. Sci. Res. 2019, 4, 763–773.
- Prasad, S.; Nigam, N.; Kalra, N.; Shukla, Y. Regulation of signaling pathways involved in lupeol induced inhibition of proliferation and induction of apoptosis in human prostate cancer cells. Mol. Carcinog. 2008, 47, 916–924.
- Liu, Y.; Bi, T.; Shen, G.; Li, Z.; Wu, G.; Wang, Z.; Qian, L.; Gao, Q. Lupeol induces apoptosis and inhibits invasion in gallbladder carcinoma GBC-SD cells by suppression of EGFR/MMP-9 signaling pathway. Cytotechnology 2014, 68, 123–133.
- Tarapore, R.S.; Siddiqui, I.A.; Saleem, M.; Adhami, V.M.; Spiegelman, V.S.; Mukhtar, H. Specific targeting of Wnt/ -catenin signaling in human melanoma cells by a dietary triterpene lupeol. Carcinogenesis 2010, 31, 1844–1853.
- Wang, Y.; Hong, D.; Qian, Y.; Tu, X.; Wang, K.; Yang, X.; Shao, S.; Kong, X.; Lou, Z.; Jin, L. Lupeol inhibits growth and migration in two human colorectal cancer cell lines by suppression of Wnt–β-catenin pathway. Onco Targets Ther. 2018, 11, 7987–7999.
- Saleem, M.; Kaur, S.; Kweon, M.-H.; Adhami, V.M.; Afaq, F.; Mukhtar, H. Lupeol, a fruit and vegetable based triterpene, induces apoptotic death of human pancreatic adenocarcinoma cells via inhibition of Ras signaling pathway. Carcinogenesis 2005, 26, 1956–1964.
- Rauth, S.; Ray, S.; Bhattacharyya, S.; Mehrotra, D.G.; Alam, N.; Mondal, G.; Nath, P.; Roy, A.; Biswas, J.; Murmu, N. Lupeol evokes anticancer effects in oral squamous cell carcinoma by inhibiting oncogenic EGFR pathway. Mol. Cell. Biochem. 2016, 417, 97–110.
- Min, T.R.; Park, H.J.; Ha, K.T.; Chi, G.Y.; Choi, Y.H.; Park, S.H. Suppression of EGFR/STAT3 activity by lupeol contributes to the induction of the apoptosis of human non-small cell lung cancer cells. Int. J. Oncol. 2019, 55, 320–330.
- Prasad, N.; Sabarwal, A.; Yadav, U.C.S.; Singh, R.P. Lupeol induces S-phase arrest and mitochondria-mediated apoptosis in cervical cancer cells. J. Biosci. 2018, 43, 249–261.
- Kangsamaksin, T.; Chaithongyot, S.; Wootthichairangsan, C.; Hanchaina, R.; Tangshewinsirikul, C.; Svasti, J. Lupeol and stigmasterol suppress tumor angiogenesis and inhibit cholangiocarcinoma growth in mice via downregulation of tumor necrosis factor-α. PLoS ONE 2017, 12, e0189628.

