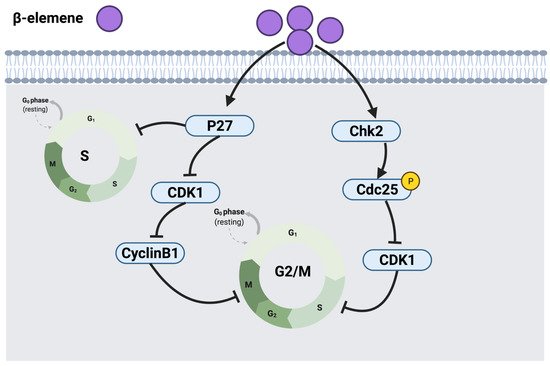
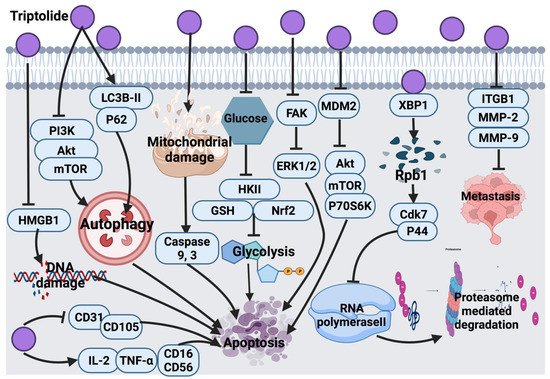
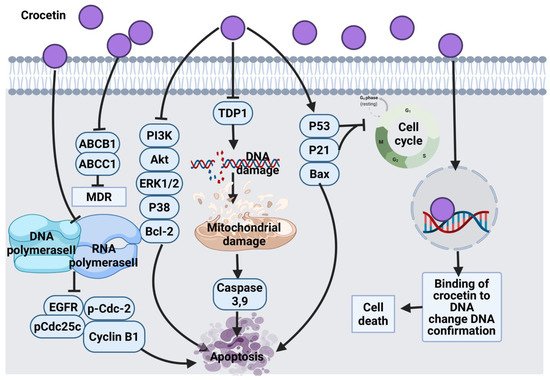
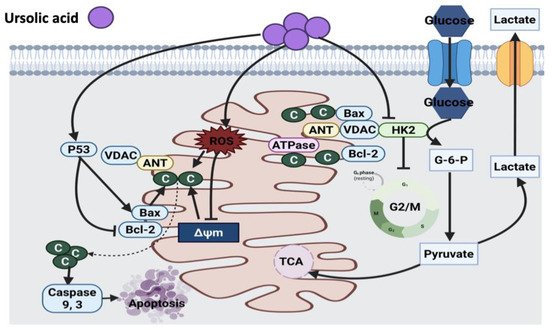
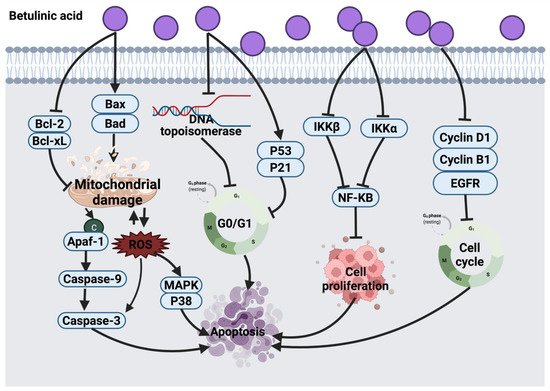
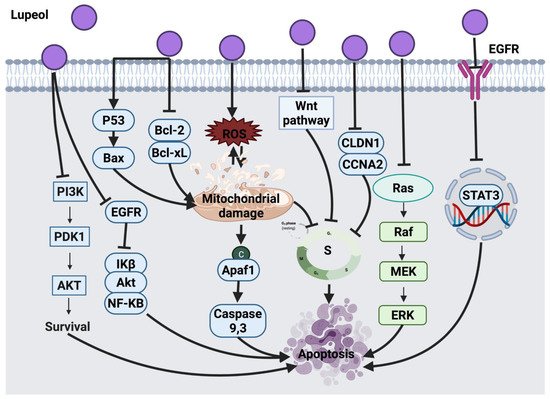
2.1.1. Thymol
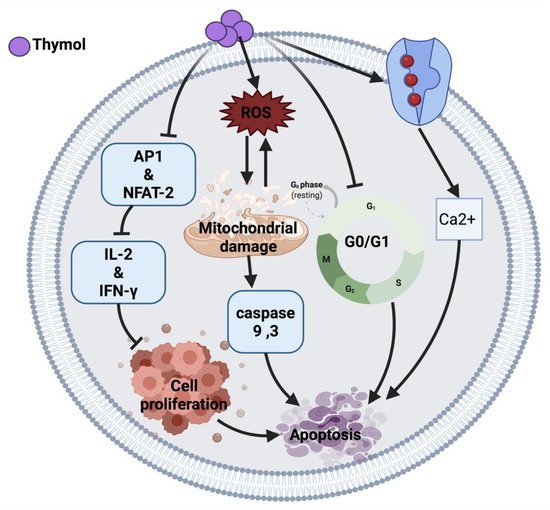
Thymol induces an anticancer effect in acute promyelocytic leukemia cells (HL-60) by arresting the cell cycle at G0/G1 phase and mitochondrial depolarization. The cytotoxic effect of thymol is through reactive oxygen species (ROS) production followed by mitochondrial membrane deterioration. Thymol also can increase the expression of Bax protein and reduce Bcl-2 protein, leading to the activation of caspases and apoptosis [
10]. Moreover, thymol induced lipid degeneration, mitochondrial depolarization, nucleolar segregation and apoptosis in Caco-2 colorectal adenocarcinoma cells. Although the ultrastructural study showed cytotoxicity of thymol against cancer cells, further studies are required to identify the mechanisms that may be involved in this cytotoxicity [
12]. A similar effect of thymol was also observed in AGC human gastric carcinoma cells, where the intrinsic apoptosis pathway and ROS generation resulted in cell death [
13].
Similarly, thymol induced apoptosis in bladder cancer cells (T24, SW780, J82) through caspases activation [
15]. Conversely, thymol in a 7-day treatment study demonstrated an anti-apoptotic response via the Bcl-2/Bax pathway [
11]. Thymol-induced mitochondrial damage was also observed in oral squamous carcinoma cells (OSCC) and cervical cancer cells in a xenograft nude mice model [
14]. In addition, thymol exerted its anticancer effect in human T lymphocyte Jurkat cells (JM) by playing an immunomodulatory role on T-cell activity via reducing interleukin-2 (IL-2) and interferon-gamma (IFN-
Y) production. The proposed mechanism of action of thymol is due to the down-regulation of activator protein 1 (AP-1) and nuclear factor of activated T-cell (NFAT-2) as a transcription factor [
16].
Thymol exerts its anticancer effect by inducing phospholipase C-dependent Ca
2+ secretion from the endoplasmic reticulum and Ca
2+ entry through store-operated Ca
2+ channels, which may activate apoptosis in MG63 human osteosarcoma cells [
17], DBTRG-05MG human glioblastoma cells [
18] and PC3 human prostate cancer cells [
19]. Moreover, thymol-induced Ca
2+ secretion induces ROS generation, thus the exact mechanism of cell death is unclear [
17].
Figure 1 illustrates an overview of the possible anticancer mechanisms of thymol.
Figure 1. Molecular mechanisms involved in the effect of thymol on cancer cells. Ca2+: calcium ion, ROS: reactive oxygen species, AP1: activator protein 1, NFAT-2: nuclear factor of activated T cell, IL-2: interleukin-2, IFN-Y: interferon-gamma.
2.1.2. Menthol
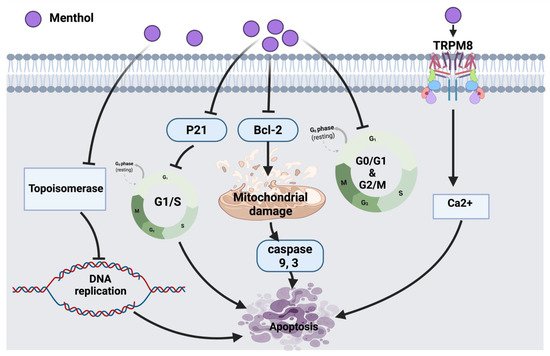
According to Lin et al., the inhibitory molecular mechanism of menthol in SNU-5 human gastric cancer cells triggered the reduction of topoisomerase I and II followed by DNA damage and cell death. Eventually, targeting the suppression of topoisomerases could be a potential therapeutic strategy to be used in combination with other clinical approaches [
20]. Another study has shown that 1alpha, 25-dihydroxyvitamin D3 (1α,25(OH)
2D
3) in combination with menthol might enhance the anticancer effect against prostate cancer cells via targeting the reduction of Bcl-2 protein expression and suppression of p21. Moreover, in other types of cancer, the 24-hydroxylase enzyme, which is a 1α,25(OH)
2D
3 catabolic enzyme, was shown to be up-regulated; this led to the inactivation of 1α,25(OH)
2D
3 and suggests a tumor escaping mechanism. Moreover, the effect of menthol on metabolic enzymes could be a challengeable approach in drug discovery [
21]. It has been reported that cancer induces the efflux of Ca
2+ from bones into the bloodstream, hence activating a calcium-permeable channel (TRPM8) resulting in cell death [
23]. Menthol, in bladder cancer cell line (T24) induced mitochondrial membrane depolarization via increasing the Ca
2+ level through the overexpression of TRPM8 in T24 cells leading to cancer cell death [
22].
Menthol also arrests cell cycles in androgen-independent DU145 prostate cancer cells at the G
0/G
1 phase by down-regulating CDK2, 4, 6 [
24]. Another study that tested PC3 prostate cancer cells showed that menthol has the potential to arrest the cell cycle at the G2/M phase by down-regulating the downstream signaling of polo-like kinase 1 (PLK1). The outcomes provide a pharmacological basis for future investigations of menthol against other cancer cells [
25]. The overall anticancer molecular mechanism of menthol is illustrated in
Figure 2.
Figure 2. Molecular mechanisms involved in the effect of menthol on cancer cells. Bcl-2: B-cell lymphoma 2, TRPM8: transient receptor potential cation channel subfamily member 8, Ca2+: calcium ion, p21: cyclin-dependent kinase inhibitor.
2.1.3. Auraptene
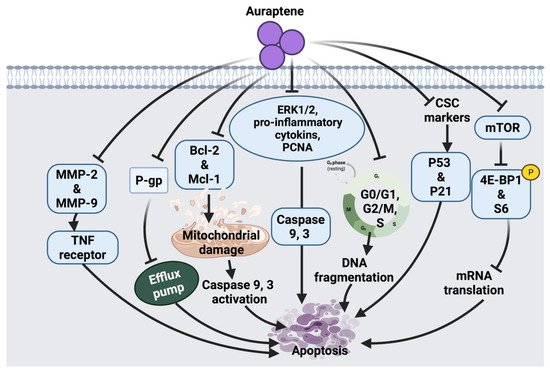
Auraptene arrests the cell cycle and induces apoptosis in SNU-1 human gastric cancer cells via mitochondrial membrane damage. Auraptene was shown to up-regulate p53 tumor suppressor protein and decrease cyclin D1 levels; therefore, arresting cell progression through the G1 phase, leading to cell death. Auraptene also inhibited cell proliferation by down-regulating the mTOR signaling pathway and its downstream activity. Although this suggested anticancer mechanism of auraptene triggered mTOR inhibition, indirect activation of mTOR-rictor is a currently unknown mechanism that could be further investigated [
27]. The synergistic role of auraptene together with anticancer drugs (cisplatin, paclitaxel, 5-fluorouracil) was studied on KYSE30 esophageal carcinoma cells. Auraptene enhanced the anticancer drug efflux transporters via interaction with the P-gp efflux pump, which increases the drug’s toxic effects inside the cancer cells. Auraptene also attenuates the malignant properties of cancer cells through activating p53 and p21 overexpression and down-regulation of cancer stem cells (CSC) markers, such as CD44 and BMI-1 [
26].
Moreover, the effect of auraptene on human cervical cancer cells (HeLa cell) and ovarian cancer cells (A2780) showed growth-inhibitory effects, suppression of cell migration and metastasis. The possible anticancer effect of auraptene in these cell lines was suggested due to a decline in the expression levels of matrix metalloproteinases (MMP-2 and MMP-9) [
28]. Auraptene also inhibits β-catenin T-cell factor (TCF) function in colorectal cancer cells and suppresses the overexpression of c-Myc proto-oncogene. In addition, auraptene treatment showed cell growth inhibition and G2/M phase arrest in Caco-2 human colorectal carcinoma and DLD-1 colorectal adenocarcinoma cells. This monoterpenoid has been shown to inhibit β-catenin/TCF activity in Caco-2 cells and enhance its activity in DLD-1 cells. This contradiction in the mechanism of auraptene action indicates that the cell growth inhibition of colorectal cancer cells by auraptene may be independent of the targeting of β-catenin-TCF signaling [
30].
Another study suggested the inhibitory effect of auraptene in azoxymethane (AOM) induced colorectal preneoplastic lesions in mice. Auraptene significantly inhibited the formation of aberrant crypt foci (cancer markers), β-catenin-accumulated crypt and cell proliferation, as well as increased apoptosis [
29]. Similarly, auraptene displayed an inhibitory effect on AOM-induced colon carcinogenesis in mice by reducing proliferating cell nuclear antigen (PCNA) and increasing DNA fragmentation. Moreover, auraptene showed the potential to ameliorate pro-inflammatory cytokines suggesting that this compound exerts its inhibitory effect against cancer through modulating inflammation. It is worth mentioning that enhancing auraptene bioavailability and improving tumor targeting is necessary to increase the chemo-preventive efficacy of auraptene [
31].
Auraptene arrests MCF-7 mammary adenocarcinoma cells in the S phase through alteration of some of the transcription genes involved in cell cycle progression, including the down-regulation of cyclin D1 protein expression level and the suppression of insulin-like growth factor-1 (IGF-1). Similarly, the chemo-preventive effect of auraptene in human breast cancer cells (MCF-7 and MDA-MB-231) was reported due to the inhibition of cyclin D1 expression and cell cycle progression. Since auraptene inhibitory effect on cell cycle triggered via several pathways, further investigation on the potential synergistic effect of this compound in combination with other chemotherapeutic agents was suggested to be carried out [
32]. Moreover, the ability of auraptene to suppress the tumor incidence and multiplicity was found to be significant at week 16 after treatment; however, toward the end of the experiment, both effects were not significant. This indicates that auraptene may delay the progression of tumor growth only during the early stages of cancer; therefore, future investigation at different time points of tumor development will provide more information regarding the anti-tumor effect of auraptene [
33]. In PC3 and DU145 cancer cells, the possible anticancer mechanism of auraptene was suggested to be through Mcl-1-mediated caspase activation. Auraptene activated caspase 9 and 3, enhanced expression of Bax and reduced expression of Bcl-2 and Mcl-1 [
34]. A summarized overview of the molecular mechanism of auraptene against cancer cells is shown in
Figure 3.
Figure 3. Molecular mechanisms involved in the effect of auraptene on cancer cells. MMP-2: matrix metalloproteinase-2, MMP-9: matrix metalloproteinase-9, TNF: tumor necrosis factor, P-gp: P-glycoprotein, Bcl-2: B-cell lymphoma 2, Mcl-1: apoptosis regulator, ERK1/2: extracellular signal-regulated kinases, PCNA: proliferating cell nuclear antigen, CSC: cancer stem cell surface, p53: tumor protein, p21: cyclin-dependent kinase inhibitor, mTOR: mammalian target of rapamycin, 4E-BP1: eukaryotic translation initiation factor 4E-binding protein 1.
2.1.4. D-Limonene
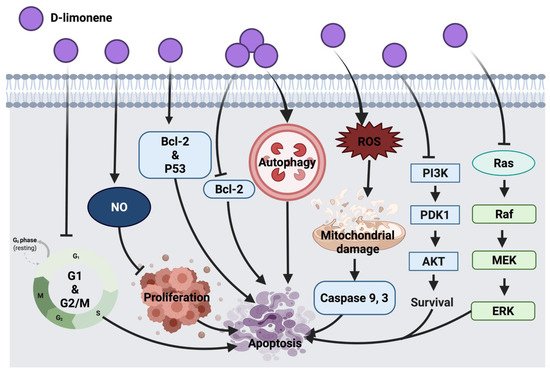
The immunomodulatory effect of D-limonene through macrophages was suggested to inhibit cell proliferation via enhancing nitric oxide (NO) production, which indicates that D-limonene anticancer activity is associated with macrophages activation in BALB/c mice with lymphoma cells. Since D-limonene was suggested to modulate immune response via macrophages, future consideration would be the investigation of mechanisms involved in macrophages activation and innate and specific immunity [
37]. D-limonene inhibited HL-60 leukemia cell proliferation through the down-regulation of Bcl-2 and alteration of the p53 gene while enhancing the expression of the pro-apoptotic Bax gene [
38]. In lung cancer (A549 and H1299), this compound exerted autophagy response via the cleavage of Atg5 pro-apoptotic agent and through the interaction with Bcl-XL transmembrane protein in mitochondria. This resulted in the activation of caspases involved in apoptosis and cell death. Partial involvement of Atg5 was speculated in D-limonene-induced apoptosis, thus a deep investigation into the mechanism underlying the role of autophagy in D-limonene-induced apoptosis could be considered in the future [
35]. Furthermore, the synergistic effect of D-limonene with berberine was evaluated in MGC803 human gastric cancer cells, where it stimulated ROS generation intracellularly, facilitated mitochondrial damage, up-regulated caspase 3 activity and down-regulated Bcl-2 protein expression and mediated cell death. Additionally, D-limonene’s synergistic effect with berberine showed cell cycle suppression at G
1 and G
2/M phases and apoptosis promotion through the intrinsic mitochondrial pathway. However, additional investigations are required to elucidate mechanistic interactions of the combined drugs [
39]. Skin tumor treatment with D-limonene inhibited the expression of Ras, Raf, MEK and ERK1/2. This anticancer mechanism was detected along with the down-regulation of Bcl-2 and up-regulation of Bax proteins [
40]. PI3K/AKT pathway inhibition was detected as an anticancer mechanism of D-limonene in LS174T colon cancer cells [
36]. Further investigation in other colon cancer cell lines could be considered in future studies.
Figure 4 represents an overview of the anticancer molecular mechanism of D-limonene.
Figure 4. Molecular mechanisms involved in the effect of D-limonene on cancer cells. Bcl-2: B-cell lymphoma 2, Bax: Bcl-2 associated X protein, p53: tumor protein, NO: nitric oxide, ROS: reactive oxygen species, PI3K: phosphoinositide 3-kinase, PDK1: 3-phosphoinositide-dependent kinase 1, Akt: protein kinase B, Ras: rat sarcoma, Raf: rapidly accelerated fibrosarcoma, MEK: mitogen-activated protein kinase, ERK: extracellular signal-regulated kinases.
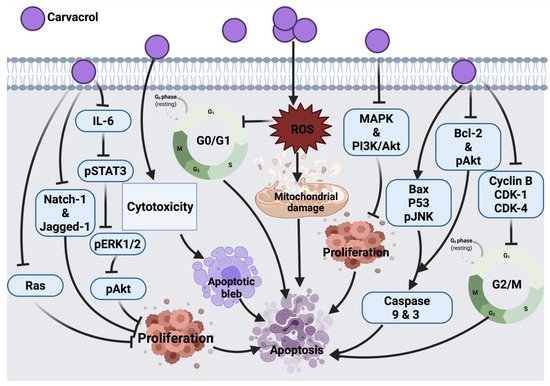
2.1.7. Carvacrol
Carvacrol exerts its tumor inhibitory effect in HCT116 human colon cancer cells by blocking MMP-2 and MMP-9, a similar mechanism exerted by auraptene on cancer cells. Treatment of HCT-116 and LoVo human colon cancer cells with this compound also blocked cyclin B1 expression and G
2/M phase progression. This compound was shown to reduce the expression level of anti-apoptotic proteins (Bcl-2 and phosphorylation of Akt) and increase apoptotic stimulating agents (Bax and JNK phosphorylation). It also showed an inhibitory role in the survival of signaling pathways, such as MAPK and PI3K/Akt; thus, suppressing cell proliferation. One of the limitations of this study is the high dosage of carvacrol used to trigger apoptosis. Indeed, 100–700 μM/L is relatively high compared to other anticancer agents [
45].
In AGS human gastric adenocarcinoma cells, carvacrol was shown to initiate apoptosis and ROS generation leading to cell death; however, only at higher concentrations (tested concentration range: 0–100 μM/L) could carvacrol exert such an effect [
46]. In MCF-7 breast cancer cells, this compound was shown to induce cell death by activating the apoptosis pathway via modulating p53, Bcl-2 and Bax proteins expression and stimulated caspases 9, 3 activations and DNA fragmentation [
55]. Another study described the similar apoptotic molecular mechanism of carvacrol in HepG-2 (human hepatocarcinoma) cells [
56]. Similar to auraptene, carvacrol in an in vivo study showed the potential to inhibit tumor development by suppressing aberrant crypt foci formation in colorectal cancer cells (CRC) through enhancing colonic lipid peroxidation and antioxidant enzymes activity. In this study, the maximum inhibitory effect against DMH-induced colon carcinogenesis was mediated by an intermediate dose of 40 mg/kg in comparison to lower (30 mg/kg) and higher (80 mg/kg) doses. At a lower dose, carvacrol concentration was not enough to alleviate DMH-induced damage, while at high concentration, carvacrol may change its antioxidant activity followed by a decrease in its potential [
57]. Therefore, a long-term study on biochemical and molecular aspects is suggested to pinpoint the main mechanism by which carvacrol induces its anticancer effect against colon carcinogenesis. The cytotoxic effect of carvacrol in A549 lung carcinoma epithelial cells was proven by targeting cytoplasmic blebs formation and DNA damage. However, this effect was shown in cells treated with 500–1000 μM/l carvacrol, which remains a high dose range compared to other anticancer drugs [
58]. In addition, carvacrol in LNCaP [
59], MDA-MB-231 and U87 [
207,
208] and HEp-2 (human epithelial type 2) [
62] cancer cells mediated cell death via mitochondrial membrane depolarization and apoptosis induction.
Moreover, carvacrol induced the down-regulation of IL-6, which resulted in the reduced expression of pSTAT3, pERK1/2 and pAkt cellular signaling proteins and resulted in the inhibition of cell proliferation, viability and invasion of PC3 cells. In this study, the authors did not investigate other possible mechanisms involved in the effect of carvacrol against PC3, such as apoptosis, necrosis, NF-
KB, cleavage of caspase 3, Bax and Bcl-2 protein expression. This could be suggested in future research [
47].
In DU145 human prostate cancer cells, carvacrol induced ROS-linked apoptosis with cell cycle suppression at G
0/G1 phase. Further study is underway to explore the molecular mechanism involved in the anticancer effect of carvacrol [
48]. A recent study showed that the inhibitory effect of carvacrol on the cell cycle is associated with the down-regulation of cyclin D1 and CDK4 and the up-regulation of suppressor protein p21 in androgen-independent PC-3 human prostate cancer. Aside from this, carvacrol blocked Notch-1 and Jagged-1 protein expression, resulting in the inhibition of the Notch signaling pathway [
49]. In another study, CO25 myoblast cells treated with carvacrol showed cell cycle arrest at the G
0/G1 phase; this suggested the apoptotic effect of carvacrol and suppression of DNA synthesis. Prenylation of Ras enables its association with plasma membrane followed by its oncogenic activity, the impairment in the prenylation of Ras was suggested to count for the anti-tumor activity of carvacrol. In addition to Ras, prenylated proteins TC21, Rho and PRL-PTP-CAAX tyrosine phosphatases are suggested to be oncogenic; therefore, these proteins could be taken as cellular targets for carvacrol in the future [
50]. In a diethylnitrosamine (DEN)-induced hepatocellular carcinoma animal model, carvacrol exerted its effect by scavenging free radicals and inhibiting lipid peroxidation, leading to prevent hepatic tumor formation. However, further investigation on the effects of carvacrol as an anticancer candidate still has a long way to go [
51].
Furthermore, a dose-dependent cytotoxic effect was also reported in the P815 mastocytoma cell line upon carvacrol treatment [
52]. Cell cycle arrest at the S phase was induced as a result of carvacrol induction in P815, CEM, K-562, MCF-7 and MCF-7 breast tumor cells. However, the link between the molecular structure and cytotoxic effect of carvacrol remains to be investigated. Moreover, the type of cells might be involved in their differential sensitivity to carvacrol. Whether P53 mutation in CEM and P-815 selected cell lines is associated with their sensitivity to carvacrol warrants further investigations [
53]. Another study that tested the carvacrol effect against DMBA-induced lung tumor in an animal model showed a significant anti-tumor effect of carvacrol; however, the mechanism of anti-tumor activity of this compound was not assessed. As a future consideration, a link between calcium metabolism and the anti-tumor effect of carvacrol was suggested [
54]. The anticancer molecular mechanism of carvacrol against different cancer cells is illustrated in
Figure 5.
Figure 5. Molecular mechanisms involved in the effect of carvacrol on cancer cells. IL-6: interleukin 6, pSTAT3: phosphorylated signal transducer and activator of transcription 3, pERK1/2: phosphorylated extracellular signal-regulated kinase 2, Natch-1 and Jagged-1: oncogenes, Ras: rat sarcoma, pAkt: phosphorylated Protein kinase B, ROS: reactive oxygen species, MAPK: mitogen-activated protein kinase, PI3K/Akt: Phosphoinositide 3-kinase/ Protein kinase B, Bax: Bcl-2 associated X protein, p53: tumor protein, pJNK: phosphorylated c-Jun N-terminal kinase, Bcl-2: B-cell lymphoma 2, CDK-1: cyclin-dependent kinase 1, CDK-4: cyclin-dependent kinase 4.
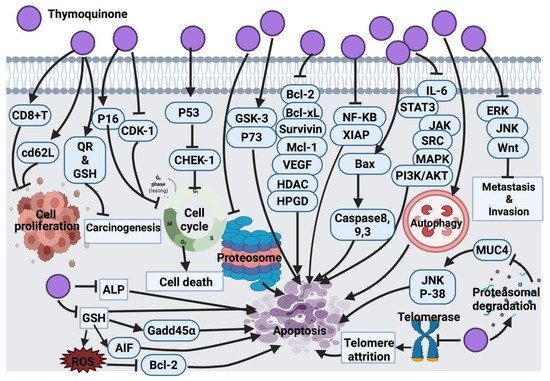
2.1.8. Thymoquinone
Findings from Rooney and collaborators [
209] revealed that HEp-2 cells are the most sensitive to the cytotoxic effect of thymoquinone compared to A-549, HT-29 and MIA PaCa-2 (pancreas carcinoma) cell lines. In their study, the anticancer mechanism of thymoquinone was demonstrated through targeted mitochondrial glutathione (GSH) depletion, activation of caspase 3 and apoptosis. Although thymoquinone-treated HEp-2 cells (25 and 50 μM/L) induced apoptosis, at higher concentrations of thymoquinone (75 μM/L), necrosis was the dominant cellular death mechanism [
209]. In another study, black cumin essential oil showed anticancer activity against A549 and DLD-1 (colon adenocarcinoma) cancer cells; this activity was claimed due to the effect of thymoquinone. However, no further investigation was conducted to determine the molecular mechanism of thymoquinone [
63]. Thymoquinone activity in malignant glioma cells (U87MG and T98G) induced apoptosis and cell death by inhibiting proteasome activity. This inhibitory effect results in an enhancement in the level of apoptotic proteins, such as p53 and Bax, leading to the activation of the apoptosis cascade. Findings suggested the very low toxicity of thymoquinone, which is worth clinical analysis and examination mainly for its potential application as an adjuvant in cancer treatment [
74]. Additionally, the inhibitory effect of thymoquinone on the NF signaling pathway and apoptosis induction was investigated and confirmed in KBM-5 leukemia cells [
85]. In general, thymoquinone induced p53-dependent checkpoint kinase-1 (CHEK-1) in HCT-116 colorectal cancer cells. Since p53 mutation accounts for >50% of colorectal cancer cases and CHEK-1 activation declines the potency of DNA damaging drugs, it was speculated that thymoquinone in combination with CHEK-1 inhibitors might be effective against colorectal cancer. However, no data was provided to report whether p53 mutants are capable of suppressing CHEK-1 differently [
93].
The activation of the signal transducer and the activator of the transcription 3 (STAT3) signaling pathway is an indication of tumor survival and cell proliferation [
94]. It has been found that STAT3 is activated through ligands binding to inflammatory cytokines (IL-6) and growth factors and initiates a downstream signaling pathway via recruitment of adaptor kinases (JAKs and SRC) [
95]. Thymoquinone has the potential to inhibit IL-6 induced STAT3 activation in multiple myeloma (MM) cells (U266 and RPMI8226), followed by blocking JAK and SRC, resulting in the suppression of the downstream signaling pathway. On the other hand, thymoquinone potentially blocked the expression of STAT3 regulatory genes, such as cyclin D1, Bcl-2, Bcl-xL, survivin, MCI-1 and VEGF [
96]. It also induced cell cycle arrest at the G1 phase and activated apoptosis. Thymoquinone, in combination with thalidomide and bortezomib, enhanced the apoptotic effect of the drugs in multiple myeloma (MM) cells through the inhibition STAT3 signaling pathway [
96]. Although thymoquinone was introduced as a potentially effective suppressor of proliferation and angiogenesis via inhibition of STAT3, a further in vivo study may highlight thymoquinone as a potential therapeutic agent in cancers harboring active STAT3.
A significant anti-tumor activity of thymoquinone against a 20-methylcholanthrene (MC)-induced fibrosarcoma tumor was detected through antioxidant activity and interference with DNA synthesis coupled with the enhancement of the detoxification process [
97]. Barron and collaborators [
98] also tested thymoquinone as an antioxidant agent in combination with selenium on osteoblast cells (MG63). This combination was successful in decreasing the cell count and elevating cellular damage via the inhibition of alkaline phosphatase (ALP) and mitochondrial glutathione, followed by caspases activation and cell death [
98]. Thymoquinone, in another study, showed significant inhibition of cell proliferation with induction of apoptosis in p53-null osteosarcoma cells (MG63) [
64]. Thymoquinone’s mechanism of action was through initiating caspase 9 and the activation of downstream caspase 3. This had suggested that thymoquinone induced apoptosis through a p53-independent mechanism because osteosarcoma malignancies with the loss of p53 activity are not responsive to a wide range of chemotherapeutic drugs. Thus, a synergistic study suggested that thymoquinone might be a supportive measurement against resistant p53-null osteosarcoma to conventional treatment [
64]. Moreover, another study suggested the antiproliferative effect of thymoquinone in osteosarcoma SaOS-2 cells via the inhibition of the NF-
KB pathway. This study suggested the use of thymoquinone as an adjuvant to conventional chemotherapy in clinical trials [
65]. The cytotoxic effect of thymoquinone on HeLa cells was reported through a p53-dependent pathway [
66]. Thymoquinone suppressed colon cancer cells proliferation (HT-29, HCT-116, DLD-1 and LoVo, Caco-2) with no inhibitory effect on normal intestinal cells (FHs74lnt). Further investigations on DLD-1 cells underlined the apoptotic cell death mechanism for the thymoquinone inhibitory effect; this effect was induced by the generation of ROS. Thymoquinone enhanced the phosphorylation of JNK and ERK (mitogen-activated protein kinases (MAPK)). These findings linked the pro-oxidant activity of thymoquinone with its apoptotic efficacy in colon cancer along with a protective role of MAPK. [
67].
At the cellular level, thymoquinone was suggested to act as a pro-oxidant upon exposure to copper and lead to ROS-induced DNA breakage and cell death. The suggested mechanism for this effect was contemplated to be a non-enzymatic copper-dependent pathway for the cytotoxic effect of thymoquinone that may be able to mobilize and reduce endogenous copper [
68]. The clinical application of CB1958 (a chemotherapy drug) is associated with hepatotoxicity. The application of thymoquinone in combination with CB1954 reduced hepatotoxicity and improved the anticancer effect of the chemotherapy in the resistant mouse mammary gland cell line (66cl-4-GFP). Indeed, thymoquinone inhibited angiogenesis, which resulted in hypoxia in tumor tissue and mediated the transformation of CB1954 into a therapeutic form. This explains the enhancement effect of thymoquinone in cancer cells treated with CB1954 in combination with thymoquinone [
69]. In continuation of the thymoquinone anticancer effect, the amplification of antioxidant delivery also targeted prostate cancer treatment in the LNCaP cell line [
70,
71]. Additionally, thymoquinone via GSH depletion induced ROS generation and growth inhibition in C4-2B and PC-3 prostate cancer cells. The findings suggested an up-regulation of Gadd45α and apoptosis-inducing factor (AIF) and down-regulation of Bcl-2 family proteins as the effect of thymoquinone in prostate cancer cells [
72]. Thymoquinone induced mitochondrial damage, caspase 8 and caspase 3 activation and apoptosis in p53 null HL-60 cells. Caspase 8 induced activation by thymoquinone has highlighted the impact of the death receptor system. To consider further investigations, it would be effective to examine whether the death receptor/ligand system (CD95 (APO-1)/Fas) is associated with thymoquinone-induced caspase 8 activations. It might also be worth investigating whether thymoquinone has any effect on the Fas-involving proteins with the death domain [
73]. In HepG2 cells, thymoquinone also induced apoptosis by activating caspases 9 and 3 and cell cycle arrest at the G1/S phase [
75].
Thymoquinone in papilloma (SP-1) cells stimulated G
0/G
1 arrest due to the enhancement of p16 expression (cyclin-dependent kinase inhibitor) and suppression of cyclin D1 expression [
76]. A similar molecular mechanism of thymoquinone was also found in the apoptosis induction of breast cancer cells (T-47D and MDA-MB-468) [
77]. In l7 carcinoma cells, thymoquinone blocked cellular growth via G
2/M phase arrest; this mechanism is associated with elevated p53 protein expression and the down-regulation of cyclin B1 protein expression. Further studies are required to focus on examining the effect of thymoquinone on other cell-cycle and apoptosis regulators in an attempt to understand the exact mechanism of action of thymoquinone [
76]. Later, the effect of thymoquinone on glioblastoma cancer cells (M059K and M059J) was revealed due to the inhibition of telomerase, which resulted in telomere attrition, DNA damage and cell death. Further research must be directed downstream of DNA-PKcs, which is necessary for external agent-induced telomere attrition in human cells [
78]. Another published study revealed a cytotoxic effect of thymoquinone on Neuro-2a mouse neuroblastoma cells via the activation of caspase 3 and the down-regulation of XIAP caspase inhibitor. However, additional studies are required to picture the genome-wide effects of thymoquinone to explore its curative potential [
79].
Aberrant expression of glycoprotein mucin 4 (MUC4) is a marker of pancreatic cancer cells (FG/COLO357). Thymoquinone down-regulated the expression of MUC4 via proteasomal degradation. This has activated apoptosis in pancreatic cancer cells through JNK signaling activation and the p38 mitogen-activated protein kinase pathway [
80]. A pre-clinical in vivo study model must be carried out to examine the therapeutic effect of thymoquinone more precisely.
Doxorubicin administration is associated with ROS generation and cardiotoxicity. Thymoquinone as an antioxidant agent reduced cardiotoxicity in mice without disturbing the anti-tumor activity of doxorubicin. This effect was proven by the alteration of serum cardiac biomarker levels as a result of thymoquinone treatment. This study suggested that thymoquinone does not interfere with the anti-tumor activity of doxorubicin rather it functions as a powerful selective cytoprotective compound, which ameliorates cardiotoxicity without reducing doxorubicin’s anticancer effect [
81]. Thymoquinone also induced protection against carcinogenesis and toxicity in mice via the induction of detoxifying enzymes, such as quinone reductase (QR) and glutathione transferase (phase 2 enzymes) [
82]. Future investigations can be focused on the evaluation of thymoquinone phase 2 enzyme’s induction in various animal species using different doses, routes and longer administration periods. Thymoquinone enhanced CD8
+T cell activity and CD62L receptor expression, which increased the expression of interferon-gamma, suggesting that thymoquinone conditioned T cells against cancer progression [
83].
The successful treatment of thymoquinone against several cancer cell lines ((lung (LNM35), liver (HepG2), colon (HT29), melanoma (MDA-MB-435), breast (MDA-MB-231 and MCF-7)) were also reported to be through the activation of intrinsic apoptosis pathway upon the suppression of Akt phosphorylation. Thymoquinone also supported the cisplatin anticancer effect in tumor cells. The inhibitory effect was associated with highly expressed caspase 3. In silico target, determination suggests that thymoquinone induced DNA damage by targeting histone deacetylase activity (HDAC) and human 15-hydroxyprostaglandin dehydrogenase (HPGD) [
84]. Another study showed the promising anticancer effect of thymoquinone was through its interference with polyp progression in ApcMin mice via the induction of tumor cell-specific apoptosis and controlling the Wnt signaling pathway by activating GSK-3 beta. This activation resulted in the accumulation of β-catenin and a decline in the expression of nuclear c-Myc [
86]. Apoptosis activation through p38 inhibition was also detected as a result of thymoquinone treatment in T28 oral cancer cells; however, there was no animal study carried out to support this finding [
87]. Thymoquinone also induced apoptosis in squamous cell carcinoma (A431, HEp-2, and RPMI 2650) via targeting caspase cascade, JNK and Akt pathways [
88]. The anticancer effects of thymoquinone on human astrocytoma cells (U87) and human T lymphocyte cell line Jurkat (leukemia cell) disclosed the degradation of alpha/beta-tubulin. This resulted in the overexpression of p73 (tumor suppressor protein) followed by apoptosis induction in cancer cells. However, apoptosis was not detected in the non-cancerous human fibroblast cells suggesting the selective activity of thymoquinone on alpha/beta-tubulin in cancer cells. Since thymoquinone is a small drug-like molecule, it is more likely this compound will decline alpha/beta-tubulin level in cancer cells via the enhancement of proteasomal degradation. Although this study reported the selective degradation of alpha/beta-tubulin proteins in astrocytoma and leukemia cells, the mechanism involved in the inhibitory effect of thymoquinone has yet to be clarified [
89].
A more recent study revealed the anticancer effect of thymoquinone via the activation of caspase 9 in glioma and prostate cancer cell lines (T98, LNCaP and 3T3) [
90]. A similar anticancer mechanism of action of thymoquinone was also detected in renal carcinoma cell lines (BFTC909, 786-O and 786-O-SI3). In addition, thymoquinone suppressed the aldehyde dehydrogenase enzyme, Nanog and Oct-4 transcription factors, Nestin (metastatic marker) and CD44, which resulted in tumor growth inhibition [
91]. Zhang et al. [
92] indicated that thymoquinone has the potential to inhibit the metastasis of human renal cancer cells (ACHN and 786-O) via induction of autophagy through the AMPK/mTOR signaling pathway. This has suggested a combination of thymoquinone with autophagy inhibitors as a future remark to increase thymoquinone-induced anticancer activity.
Figure 6 represents a summarized view of the anticancer mechanism of thymoquinone.
Figure 6. Molecular mechanisms involved in the effect of thymoquinone on cancer cells. CD8+T: killer T cell, cd62L: L-selectin, QR: quinone reductase, GSH: glutathione, ALP: alkaline phosphatase, AIF: apoptosis inducing factor, Bcl-2: B-cell lymphoma 2, Gadd45α: growth arrest and DNA damage inducible alpha, p53: tumor protein, CHEK-1: checkpoint kinase-1, GSK-3: glycogen synthase kinase-3, p73: tumor protein 73, Bcl-xL: B-cell lymphoma-extra large, Mcl-1: Mcl-1 apoptosis regulator, VEGF: vascular endothelial growth factor, HDAC: histone deacetylases, HPGD: 15-hydroxyprostaglandin dehydrogenase, NF-KB: Nuclear factor kappa light chain enhancer of activated B cells, XIAP: X-linked inhibitor of apoptosis protein, Bax: Bcl-2 associated X protein, IL-6: interleukin-6, STAT3: signal transducer and activator of transcription 3, JAK: Janus kinase, SRC: oncogene, MAPK: mitogen activated protein kinase, PI3K/Akt: Phosphoinositide 3-kinase/ Protein kinase B, ERK: extra cellular signal-regulated kinase, JNK: c-Jun N-terminal kinase, Wnt: wingless related integration site, MUC4: mucin 4, p38: mitogen activated protein kinase, ROS: reactive oxygen species.