 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chiara Bellocchi | + 2093 word(s) | 2093 | 2022-03-01 07:58:12 | | | |
| 2 | Jessie Wu | Meta information modification | 2093 | 2022-03-07 01:47:04 | | |
Video Upload Options
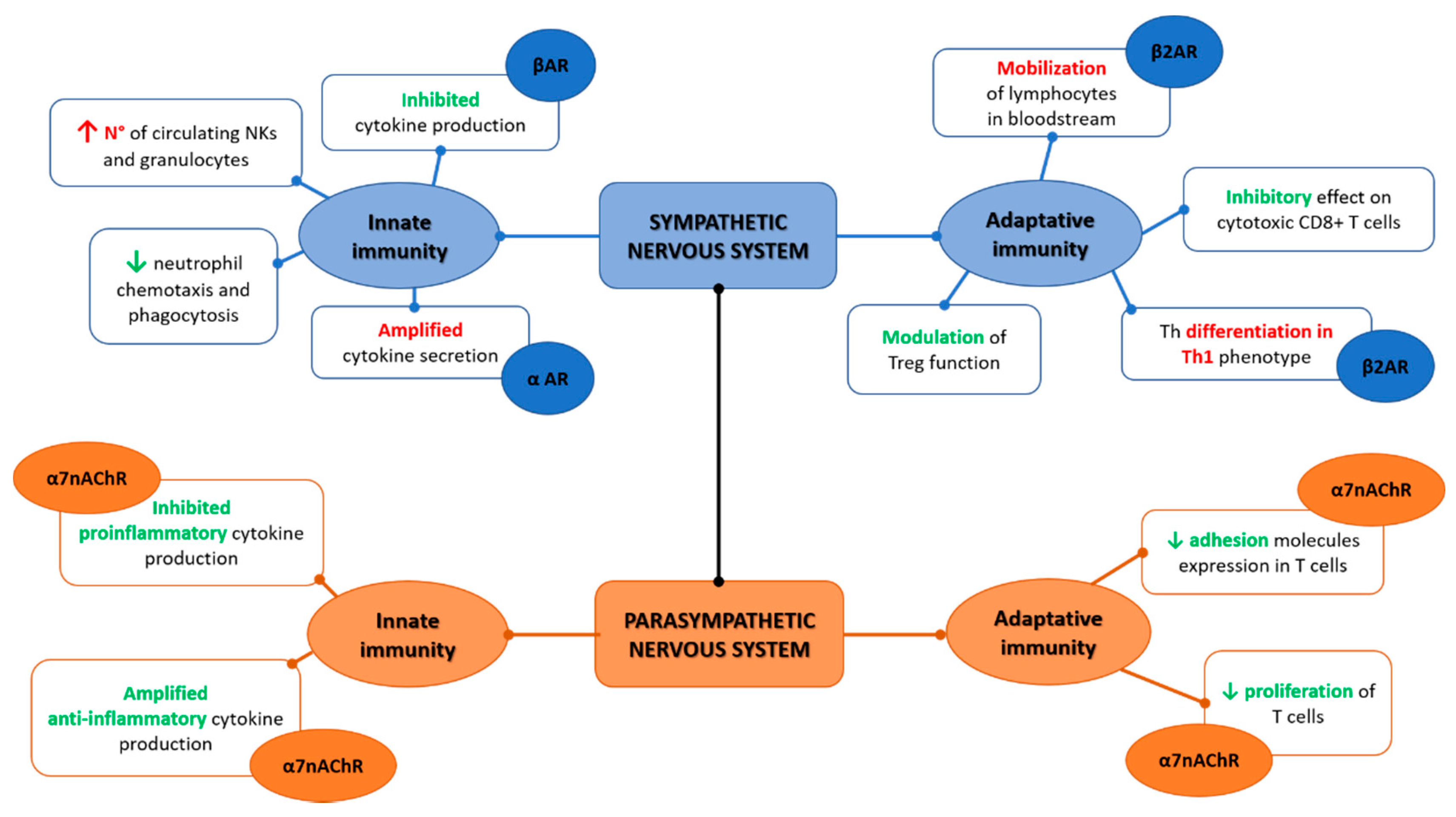
The autonomic nervous system (ANS) and the immune system are deeply interrelated. The ANS regulates both innate and adaptive immunity through the sympathetic and parasympathetic branches, and an imbalance in this system can determine an altered inflammatory response as typically observed in chronic conditions such as systemic autoimmune diseases. Rheumatoid arthritis, systemic lupus erythematosus, and systemic sclerosis all show a dysfunction of the ANS that is mutually related to the increase in inflammation and cardiovascular risk. Moreover, an interaction between ANS and the gut microbiota has direct effects on inflammation homeostasis. Recently vagal stimulation techniques have emerged as an unprecedented possibility to reduce ANS dysfunction, especially in chronic diseases characterized by pain and a decreased quality of life as well as in chronic inflammation.
1. Introduction
2. Autonomic Nervous System and Innate Immunity
3. Autonomic Nervous System and Adaptive Immunity
4. Autonomic Nervous System and Gut Microbiota
References
- Sanvictores, T.; Tadi, P. Neuroanatomy, Autonomic Nervous System Visceral Afferent Fibers and Pain; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022.
- Koopman, F.A.; Van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. Balancing the autonomic nervous system to reduce inflammation in rheumatoid arthritis. J. Intern. Med. 2017, 282, 64–75.
- Thanou, A.; Stavrakis, S.; Dyer, J.W.; Munroe, M.E.; James, J.A.; Merrill, J.T. Impact of heart rate variability, a marker for cardiac health, on lupus disease activity. Arthritis Res. Ther. 2016, 18, 197.
- Gigante, A.; Rosato, E.; Liberatori, M.; Barbano, B.; Cianci, R.; Gasperini, M.; Sardo, L.; Marra, A.; Amoroso, A.; Salsano, F.; et al. Autonomic dysfunction in patients with systemic sclerosis: Correlation with intrarenal arterial stiffness. Int. J. Cardiol. 2014, 177, 578–580.
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820.
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353.
- Shortman, K.; Caux, C. Dendritic Cell Development: Multiple Pathways to Nature’s Adjuvants. Stem Cells 1997, 15, 409–419.
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252.
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852.
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309.
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Aguilar, S.V.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; González, F.Z.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670.
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916–3924.
- Tang, S.-C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.-G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803.
- Klein, M.; Obermaier, B.; Angele, B.; Pfister, H.; Wagner, H.; Koedel, U.; Kirschning, C.J. Innate Immunity to Pneumococcal Infection of the Central Nervous System Depends on Toll-Like Receptor (TLR) 2 and TLR4. J. Infect. Dis. 2008, 198, 1028–1036.
- Zengeler, K.E.; Lukens, J.R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 2021, 21, 454–468.
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987–2007). Brain, Behav. Immun. 2007, 21, 736–745.
- Romeo, H.E.; Fink, T.; Yanaihara, N.; Weihe, E. Distribution and relative proportions of neuropeptide Y- and proenkephalin-containing noradrenergic neurones in rat superior cervical ganglion: Separate projections to submaxillary lymph nodes. Peptides 1994, 15, 1479–1487.
- Trotter, R.N.; Stornetta, R.L.; Guyenet, P.G.; Roberts, M.R. Transneuronal mapping of the CNS network controlling sympathetic outflow to the rat thymus. Auton. Neurosci. 2007, 131, 9–20.
- Cano, G.; Sved, A.F.; Rinaman, L.; Rabin, B.S.; Card, J.P. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J. Comp. Neurol. 2001, 439, 1–18.
- Bulay, O.; Mlrvish, S.S.; Pelfrene, A.F.; Eagen, M.; Garcia, H.; Gold, B. Carcinogenicity Test of Six Nitrosamides and a Nitrosocyanamide Administered Orally to Rats2. JNCI: J. Natl. Cancer Inst. 1979, 62, 1523–1528.
- Bratton, B.O.; Martelli, D.; McKinley, M.J.; Trevaks, D.; Anderson, C.R.; McAllen, R.M. Neural regulation of inflammation: No neural connection from the vagus to splenic sympathetic neurons. Exp. Physiol. 2012, 97, 1180–1185.
- Bellinger, D.; Lorton, D.; Hamill, R.; Felten, S.; Felten, D. Acetylcholinesterase Staining and Choline Acetyltransferase Activity in the Young Adult Rat Spleen: Lack of Evidence for Cholinergic Innervation. Brain, Behav. Immun. 1993, 7, 191–204.
- Sanders, V.M.; Straub, R.H. Norepinephrine, the β-Adrenergic Receptor, and Immunity. Brain, Behav. Immun. 2002, 16, 290–332.
- Sanders, V.M.; E Munson, A. Norepinephrine and the antibody response. Pharmacol. Rev. 1985, 37, 229–248.
- Daaka, Y.; Luttrell, L.; Lefkowitz, R.J. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91.
- Meltzer, J.C.; MacNeil, B.J.; Sanders, V.; Pylypas, S.; Jansen, A.H.; Greenberg, A.H.; Nance, D.M. Stress-induced suppression of in vivo splenic cytokine production in the rat by neural and hormonal mechanisms. Brain, Behav. Immun. 2004, 18, 262–273.
- Ignatowski, T.; Gallant, S.; Spengler, R.N. Temporal regulation by adrenergic receptor stimulation of macrophage (MΦ)-derived tumor necrosis factor (TNF) production post-LPS challenge. J. Neuroimmunol. 1996, 65, 107–117.
- Hetier, E.; Ayala, J.; Bousseau, A.; Prochiantz, A. Modulation of interleukin-1 and tumor necrosis factor expression by ?-adrenergic agonists in mouse ameboid microglial cells. Exp. Brain Res. 1991, 86.
- van der Poll, T.; Jansen, J.; Endert, E.; Sauerwein, H.P.; van Deventer, S.J. Noradrenaline inhibits lipopolysaccharide-induced tumor necrosis factor and interleukin 6 production in human whole blood. Infect. Immun. 1994, 62, 2046–2050.
- Severn, A.; Rapson, N.T.; A Hunter, C.; Liew, F.Y. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J. Immunol. 1992, 148.
- A Ottaway, C. Central nervous system influences on lymphocyte migration. Brain, Behav. Immun. 1992, 6, 97–116.
- Benschop, R.J.; Rodriguez-Feuerhahn, M.; Schedlowski, M. Catecholamine-Induced Leukocytosis: Early Observations, Current Research, and Future Directions. Brain Behav. Immun. 1996, 10, 77–91.
- Bellinger, D.L.; Lorton, D. Autonomic regulation of cellular immune function. Auton. Neurosci. 2014, 182, 15–41.
- Nicholls, A.J.; Wen, S.W.; Hall, P.; Hickey, M.; Wong, C.H.Y. Activation of the sympathetic nervous system modulates neutrophil function. J. Leukoc. Biol. 2017, 103, 295–309.
- Harvath, L.; Robbins, J.D.; A Russell, A.; Seamon, K.B. cAMP and human neutrophil chemotaxis. Elevation of cAMP differentially affects chemotactic responsiveness. J. Immunol. 1991, 146, 224–232.
- Zurier, R.B.; Weissmann, G.; Hoffstein, S.; Kammerman, S.; Tai, H.H. Mechanisms of Lysosomal Enzyme Release from Human Leukocytes II. EFFECTS OF cAMP AND cGMP, AUTONOMIC AGONISTS, AND AGENTS WHICH AFFECT MICROTUBULE FUNCTION. J. Clin. Investig. 1974, 53, 297–309.
- Wong, C.H.Y.; Jenne, C.N.; Lee, W.-Y.; Léger, C.; Kubes, P. Functional Innervation of Hepatic iNKT Cells Is Immunosuppressive Following Stroke. Science 2011, 334, 101–105.
- Irwin, M. Stress-induced immune suppression: Role of brain corticotropin releasing hormone and autonomic nervous system mechanisms. Adv. Neuroimmunol. 1994, 4, 29–47.
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve--an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638.
- Shakhar, G.; Ben-Eliyahu, S. In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J. Immunol. 1998, 160, 3251–3258.
- Suberville, S.; Bellocq, A.; Fouqueray, B.; Philippe, C.; Lantz, O.; Perez, J.; Baud, L. Regulation of interleukin-10 production by β-adrenergic agonists. Eur. J. Immunol. 1996, 26, 2601–2605.
- Németh, Z.H.; Szabó, C.; Haskó, G.; Salzman, A.L.; Vizi, E. Effect of the phosphodiesterase III inhibitor amrinone on cytokine and nitric oxide production in immunostimulated J774.1 macrophages. Eur. J. Pharmacol. 1997, 339, 215–221.
- Kox, M.; van Eijk, L.T.; Zwaag, J.; Wildenberg, J.V.D.; Sweep, F.; van der Hoeven, J.G.; Pickkers, P. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7379–7384.
- Kox, M.; Pickkers, P. Modulation of the Innate Immune Response through the Vagus Nerve. Nephron Exp. Nephrol. 2015, 131, 79–84.
- Gaykema, R.P.; Dijkstra, I.; Tilders, F.J. Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology 1995, 136, 4717–4720.
- Fleshner, M.; Goehler, L.; Schwartz, B.; McGorry, M.; Martin, D.; Maier, S.; Watkins, L. Thermogenic and corticosterone responses to intravenous cytokines (IL-1β and TNF-α) are attenuated by subdiaphragmatic vagotomy. J. Neuroimmunol. 1998, 86, 134–141.
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.; Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 2006, 203, 1623–1628.
- Goehler, L.E.; Relton, J.K.; Dripps, D.; Kiechle, R.; Tartaglia, N.; Maier, S.F.; Watkins, L.R. Vagal Paraganglia Bind Biotinylated Interleukin-1 Receptor Antagonist: A Possible Mechanism for Immune-to-Brain Communication. Brain Res. Bull. 1997, 43, 357–364.
- van Westerloo, D.J. The vagal immune reflex: A blessing from above. Wien. Med. Wochenschr. 2010, 160, 112–117.
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388.
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462.
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013.
- Nance, D.M.; Burns, J. Innervation of the spleen in the rat: Evidence for absence of afferent innervation. Brain Behav. Immun. 1989, 3, 281–290.
- Hosoi, T.; Okuma, Y.; Matsuda, T.; Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. 2005, 120, 104–107.
- Vida, G.; Peña, G.; Deitch, E.A.; Ulloa, L. α7-Cholinergic Receptor Mediates Vagal Induction of Splenic Norepinephrine. J. Immunol. 2011, 186, 4340–4346.
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859.
- Pontet, J.; Contreras, P.; Curbelo, A.; Medina, J.; Noveri, S.; Bentancourt, S.; Migliaro, E.R. Heart rate variability as early marker of multiple organ dysfunction syndrome in septic patients. J. Crit. Care 2003, 18, 156–163.
- Pavlov, V.A.; Ochani, M.; Yang, L.-H.; Gallowitsch-Puerta, M.; Ochani, K.; Lin, X.; Levi, J.; Parrish, W.R.; Rosas-Ballina, M.; Czura, C.J.; et al. Selective α7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis*. Crit. Care Med. 2007, 35, 1139–1144.
- Koh, D.-R.; Fung-Leung, W.-P.; Ho, A.; Gray, D.; Acha-Orbea, H.; Mak, T.-W. Less Mortality but More Relapses in Experimental Allergic Encephalomyelitis in CD8 -/- Mice. Science 1992, 256, 1210–1213.
- Bernik, T.R.; Friedman, S.G.; Ochani, M.; DiRaimo, R.; Susarla, S.; Czura, C.J.; Tracey, K.J. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J. Vasc. Surg. 2002, 36, 1231–1236.
- Dimitrov, S.; Lange, T.; Born, J. Selective Mobilization of Cytotoxic Leukocytes by Epinephrine. J. Immunol. 2009, 184, 503–511.
- Guereschi, M.G.; Araujo, L.P.; Maricato, J.T.; Takenaka, M.C.; Nascimento, V.M.; Vivanco, B.C.; Reis, V.O.; Keller, A.C.; Brum, P.C.; Basso, A.S. Beta2-adrenergic receptor signaling in CD4+Foxp3+regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur. J. Immunol. 2013, 43, 1001–1012.
- Sanders, V.M. The beta2-adrenergic receptor on T and B lymphocytes: Do we understand it yet? Brain, Behav. Immun. 2012, 26, 195–200.
- Wirth, T.; Westendorf, A.M.; Bloemker, D.; Wildmann, J.; Engler, H.; Mollerus, S.; Wadwa, M.; Schäfer, M.K.-H.; Schedlowski, M.; del Rey, A. The sympathetic nervous system modulates CD4+Foxp3+ regulatory T cells via noradrenaline-dependent apoptosis in a murine model of lymphoproliferative disease. Brain, Behav. Immun. 2014, 38, 100–110.
- Kalinichenko, V.V.; Mokyr, M.B.; Graf, L.H.; Cohen, R.L.; A Chambers, D. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J. Immunol. 1999, 163, 2492–2499.
- Livnat, S.; Madden, K.S.; Felten, D.L.; Felten, S.Y. Regulation of the immune system by sympathetic neural mechanisms. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1987, 11, 145–152.
- Kohm, A.P.; Sanders, V.M. Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo. J. Immunol. 1999, 162, 5299–5308.
- Kohm, A.P.; Mozaffarian, A.; Sanders, V.M. B Cell Receptor- and β2-Adrenergic Receptor-Induced Regulation of B7-2 (CD86) Expression in B Cells. J. Immunol. 2002, 168, 6314–6322.
- Kasprowicz, D.J.; Kohm, A.P.; Berton, M.T.; Chruscinski, A.J.; Sharpe, A.H.; Sanders, V.M. Stimulation of the B Cell Receptor, CD86 (B7-2), and the β2-Adrenergic Receptor Intrinsically Modulates the Level of IgG1 and IgE Produced per B Cell. J. Immunol. 2000, 165, 680–690.
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-Synthesizing T Cells Relay Neural Signals in a Vagus Nerve Circuit. Science 2011, 334, 98–101.
- Geng, Y.; Savage, S.; Johnson, L.; Seagrave, J.; Sopori, M. Effects of Nicotine on the Immune Response. I. Chronic Exposure to Nicotine Impairs Antigen Receptor-Mediated Signal Transduction in Lymphocytes. Toxicol. Appl. Pharmacol. 1995, 135, 268–278.
- Kawashima, K. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 86, 29–48.
- Vernino, S.; Stiles, L.E. Autoimmunity in postural orthostatic tachycardia syndrome: Current understanding. Auton. Neurosci. 2018, 215, 78–82.
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune Basis for Postural Tachycardia Syndrome. J. Am. Hear. Assoc. 2014, 3, e000755.
- Deng, J.; Li, H.; Guo, Y.; Zhang, G.; Fischer, H.; Stavrakis, S.; Yu, X. Transcutaneous vagus nerve stimulation attenuates autoantibody-mediated cardiovagal dysfunction and inflammation in a rabbit model of postural tachycardia syndrome. J. Interv. Card. Electrophysiol. 2022, 1–10.
- Molina, P.E. Noradrenergic inhibition of TNF upregulation in hemorrhagic shock. Neuroimmunomodulation 2001, 9, 125–133.
- Woiciechowsky, C.; Asadullah, K.; Nestler, D.; Eberhardt, B.; Platzer, C.; Schöning, B.; Glöckner, F.; Lanksch, W.R.; Volk, H.-D.; Döcke, W.-D. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat. Med. 1998, 4, 808–813.
- Dhabhar, F.S. Enhancing versus Suppressive Effects of Stress on Immune Function: Implications for Immunoprotection versus Immunopathology. Allergy, Asthma Clin. Immunol. 2008, 4, 2–11.
- Pongratz, G.; Straub, R.H. The sympathetic nervous response in inflammation. Arthritis Res. Ther. 2014, 16, 1–12.
- Voisine, J.; Abadie, V. Interplay between Gluten, HLA, Innate and Adaptive Immunity Orchestrates the Development of Coeliac Disease. Front. Immunol. 2021, 12.
- Cervio, E.; Volta, U.; Verri, M.; Boschi, F.; Pastoris, O.; Granito, A.; Barbara, G.; Parisi, C.; Felicani, C.; Tonini, M.; et al. Sera of Patients With Celiac Disease and Neurologic Disorders Evoke a Mitochondrial-Dependent Apoptosis In Vitro. Gastroenterology 2007, 133, 195–206.
- Volta, U.; De Giorgio, R.; Granito, A.; Stanghellini, V.; Barbara, G.; Avoni, P.; Liguori, R.; Petrolini, N.; Fiorini, E.; Montagna, P. Anti-ganglioside antibodies in coeliac disease with neurological disorders. Dig. Liver Dis. 2006, 38, 183–187.
- Kayali, S.; Selbuz, S. Assessment of Autonomic Nervous System in Children with Celiac Disease: A Heart Rate Variability Study. Indian Pediatr. 2020, 57, 719–722.
- Przybylska-Felus, M.; Furgala, A.; Zwolinska-Wcislo, M.; Mazur, M.; Widera, A.; Thor, P.; Mach, T. Disturbances of autonomic nervous system activity and diminished response to stress in patients with celiac disease. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2014, 65.
- Guy-Grand, D.; DiSanto, J.P.; Henchoz, P.; Malassis-Séris, M.; Vassalli, P. Small bowel enteropathy: Role of intraepithelial lymphocytes and of cytokines (IL-12, IFN-gamma, TNF) in the induction of epithelial cell death and renewal. Eur. J. Immunol. 1998, 28, 730–744.
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904.
- Sprockett, D.; Fukami, T.; Relman, D.A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 197–205.
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007, 5, e177.
- Umesaki, Y.; Setoyama, H.; Matsumoto, S.; Okada, Y. Expansion of alpha beta T-cell receptor-bearing intestinal intraepithelial lymphocytes after microbial colonization in germ-free mice and its independence from thymus. Immunology 1993, 79, 32–37.
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72.
- Bellocchi, C.; Volkmann, E.R. Update on the Gastrointestinal Microbiome in Systemic Sclerosis. Curr. Rheumatol. Rep. 2018, 20, 49.
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Doré, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242.
- Pozuelo, M.; Panda, S.; Santiago, A.; Mendez, S.; Accarino, A.; Santos, J.; Guarner, F.; Azpiroz, F.; Manichanh, C. Reduction of butyrate- and methane-producing microorganisms in patients with Irritable Bowel Syndrome. Sci. Rep. 2015, 5, 12693.
- Bellocchi, C.; Fernández-Ochoa, Á.; Montanelli, G.; Vigone, B.; Santaniello, A.; Milani, C.; Quirantes-Piné, R.; Borrás-Linares, I.; Ventura, M.; Segura-Carrettero, A.; et al. Microbial and metabolic multi-omic correlations in systemic sclerosis patients. Ann. N. Y. Acad. Sci. 2018, 1421, 97–109.
- Chen, B.; Jia, X.; Xu, J.; Zhao, L.; Ji, J.; Wu, B.; Ma, Y.; Li, H.; Zuo, X.; Pan, W.; et al. An Autoimmunogenic and Proinflammatory Profile Defined by the Gut Microbiota of Patients With Untreated Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020, 73, 232–243.
- Tait, C.; Sayuk, G.S. The Brain-Gut-Microbiotal Axis: A framework for understanding functional GI illness and their therapeutic interventions. Eur. J. Intern. Med. 2021, 84, 1–9.
- Mayer, E.A. Gut feelings: The emerging biology of gut–brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466.
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut Microbes and the Brain: Paradigm Shift in Neuroscience. J. Neurosci. 2014, 34, 15490–15496.
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148.
- Mayer, E.A.; Tillisch, K. The Brain-Gut Axis in Abdominal Pain Syndromes. Annu. Rev. Med. 2011, 62, 381–396.
- Berman, S.M.; Naliboff, B.D.; Suyenobu, B.; Labus, J.S.; Stains, J.; Ohning, G.; Kilpatrick, L.; Bueller, J.A.; Ruby, K.; Jarcho, J.; et al. Reduced Brainstem Inhibition during Anticipated Pelvic Visceral Pain Correlates with Enhanced Brain Response to the Visceral Stimulus in Women with Irritable Bowel Syndrome. J. Neurosci. 2008, 28, 349–359.
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2010, 23, 255-e119.
- Kelly, J.; Borre, Y.; Brien, C.O.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118.
- Bonaz, B.; Sinniger, V.; Pellissier, S. The Vagus Nerve in the Neuro-Immune Axis: Implications in the Pathology of the Gastrointestinal Tract. Front. Immunol. 2017, 8, 1452.
- Furness, J.B. Integrated Neural and Endocrine Control of Gastrointestinal Function. Enteric Nerv. Syst. 2016, 891, 159–173.
- Diepenbroek, C.; Quinn, D.; Stephens, R.; Zollinger, B.; Anderson, S.; Pan, A.; De Lartigue, G. Validation and characterization of a novel method for selective vagal deafferentation of the gut. Am. J. Physiol. Liver Physiol. 2017, 313, G342–G352.
- Powell, N.; Walker, M.M.; Talley, N.J. The mucosal immune system: Master regulator of bidirectional gut–brain communications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 143–159.
- Bartley, A.; Yang, T.; Arocha, R.; Malphurs, W.L.; Larkin, R.; Magee, K.L.; Vickroy, T.W.; Zubcevic, J. Increased Abundance of Lactobacillales in the Colon of Beta-Adrenergic Receptor Knock Out Mouse Is Associated With Increased Gut Bacterial Production of Short Chain Fatty Acids and Reduced IL17 Expression in Circulating CD4+ Immune Cells. Front. Physiol. 2018, 9.

