 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexej Dick | + 2197 word(s) | 2197 | 2022-02-23 04:11:46 | | | |
| 2 | Peter Tang | -39 word(s) | 2158 | 2022-03-04 04:12:52 | | |
Video Upload Options
Bioisosteric replacement is a powerful tool for modulating the drug-like properties, toxicity, and chemical space of experimental therapeutics. The use of bioisosteres and the introduction of structural changes to the lead compound allows the chemist to alter the compound’s size, shape, electronic distribution, polarizability, dipole, polarity, lipophilicity, and pKa, while still retaining potent target engagement.
1. Introduction
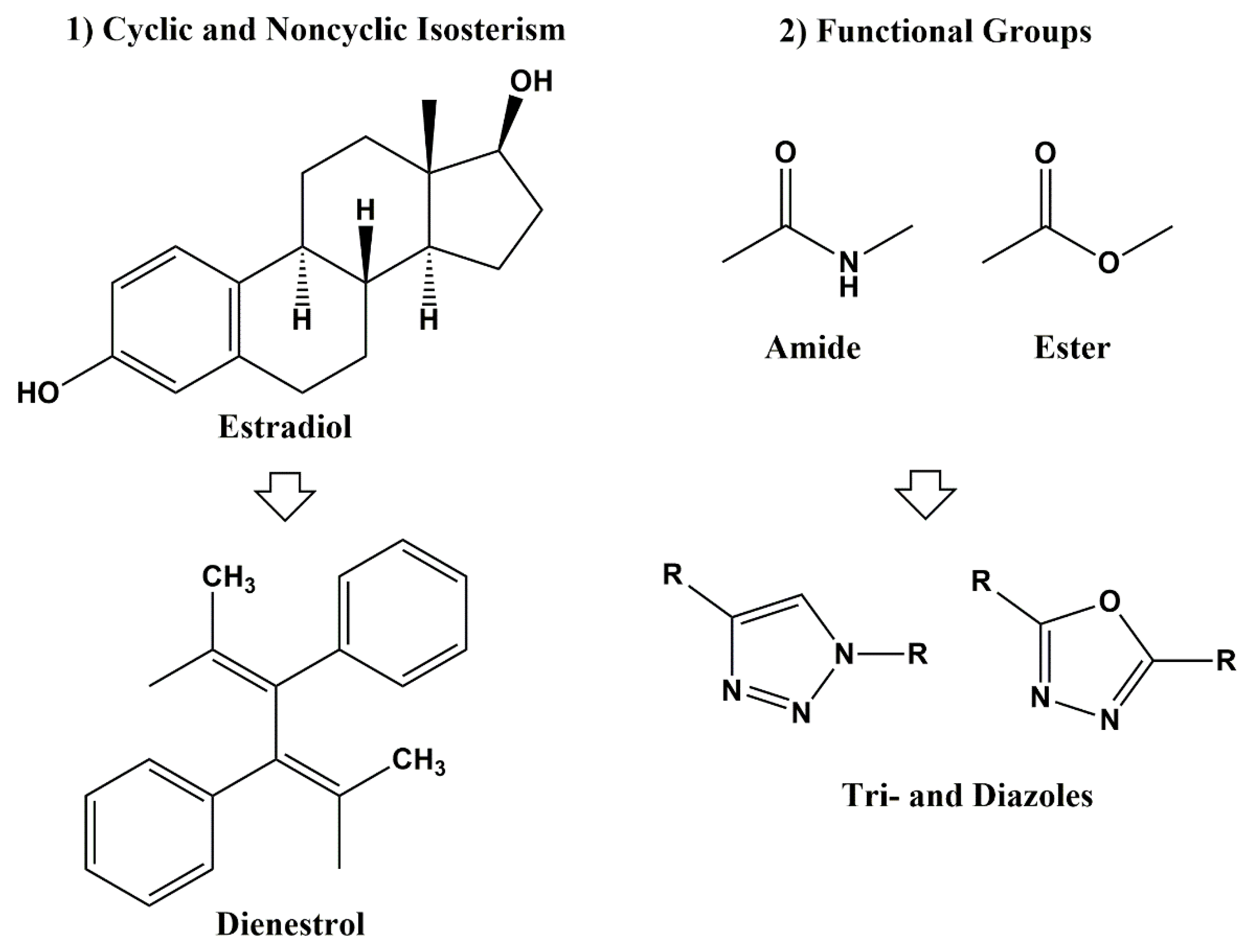
2. Principle of Bioisosterism and Historical Background
|
C |
N |
O |
F |
Ne |
Na+ |
|---|---|---|---|---|---|
|
CH |
NH |
OH |
FH |
- |
|
|
CH2 |
NH2 |
OH2 |
FH2+ |
||
|
CH3 |
NH3 |
OH3+ |
|||
|
CH4 |
NH4+ |
|
Number of Valence Electrons |
||||
|---|---|---|---|---|
|
4 |
5 |
6 |
7 |
8 |
|
N+ |
P |
S |
Cl |
ClH |
|
P+ |
As |
Se |
Br |
BrH |
|
S+ |
Sb |
Te |
I |
IH |
|
As+ |
PH |
SH |
SH2 |
|
|
Sb+ |
PH2 |
PH3 |
||
3. Classical and Non-Classical Bioisosteres
4. In Silico Molecular Field-Based Scaffold Hopping for Non-Classical Bioisostere Identification
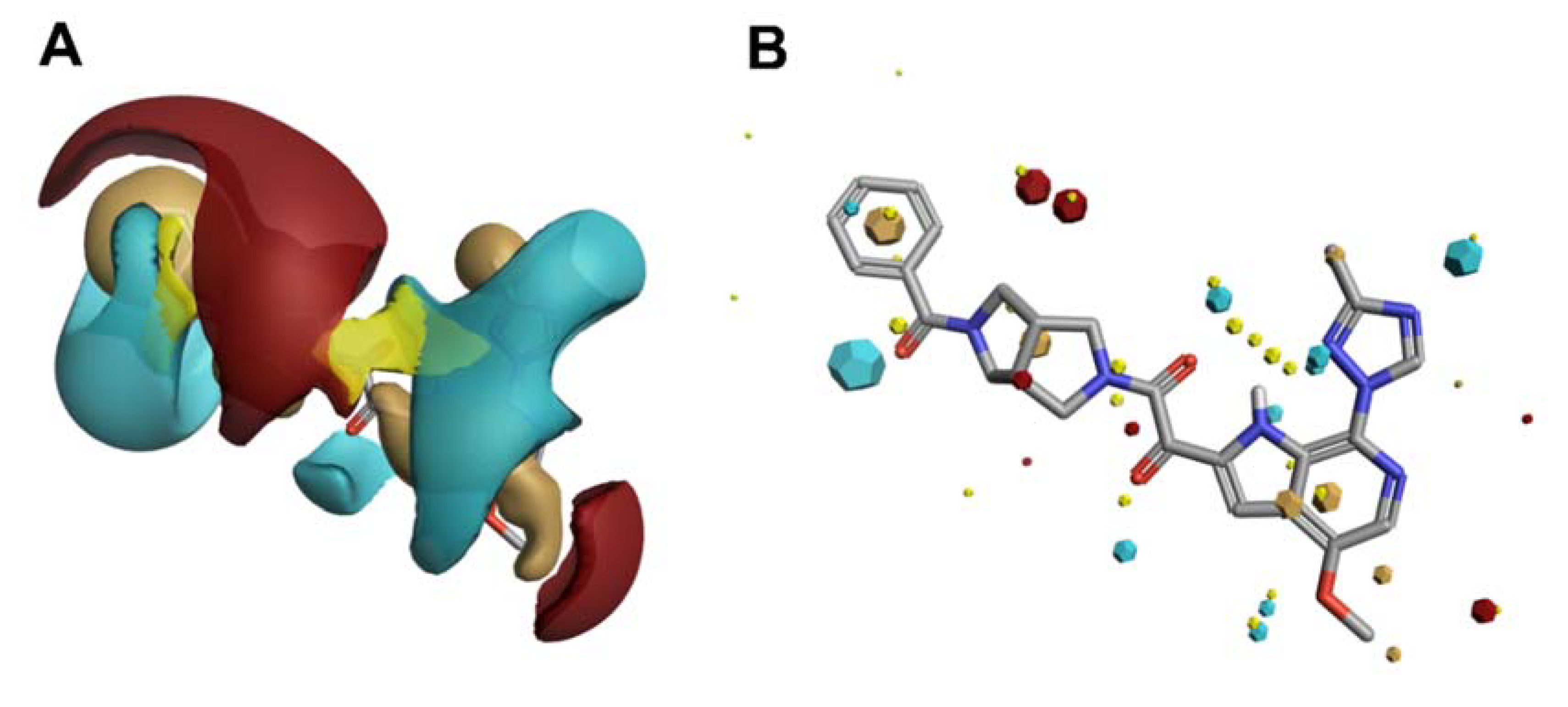
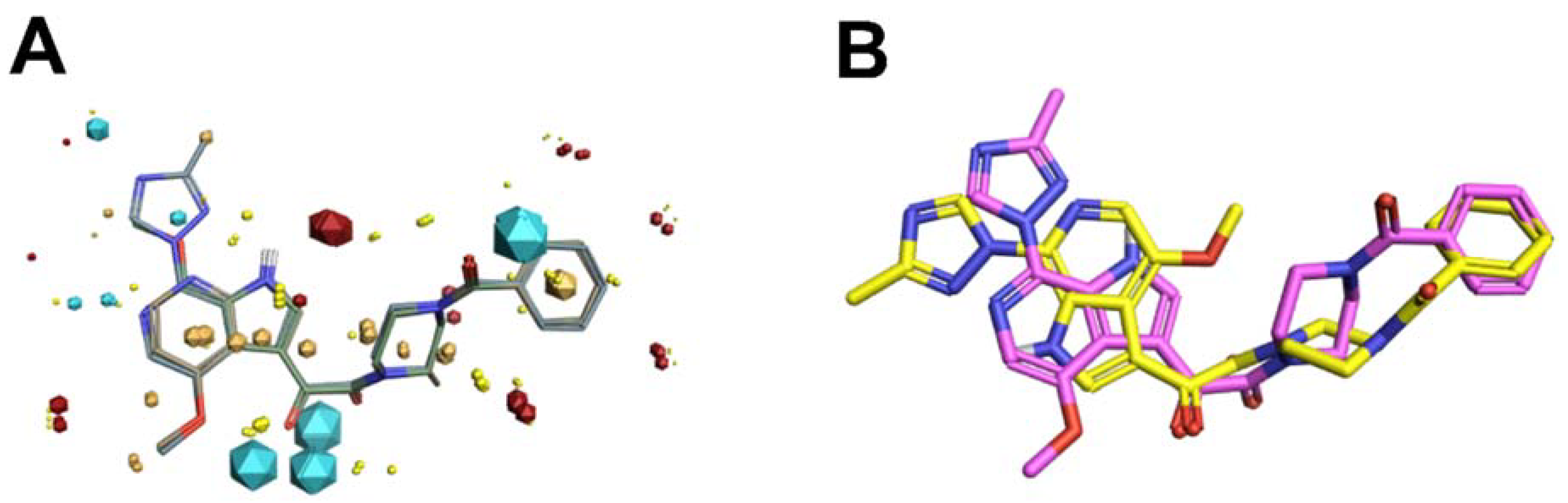
Scaffold Hopping for Potency and ADME Improvement of HIV-1 Entry Leads
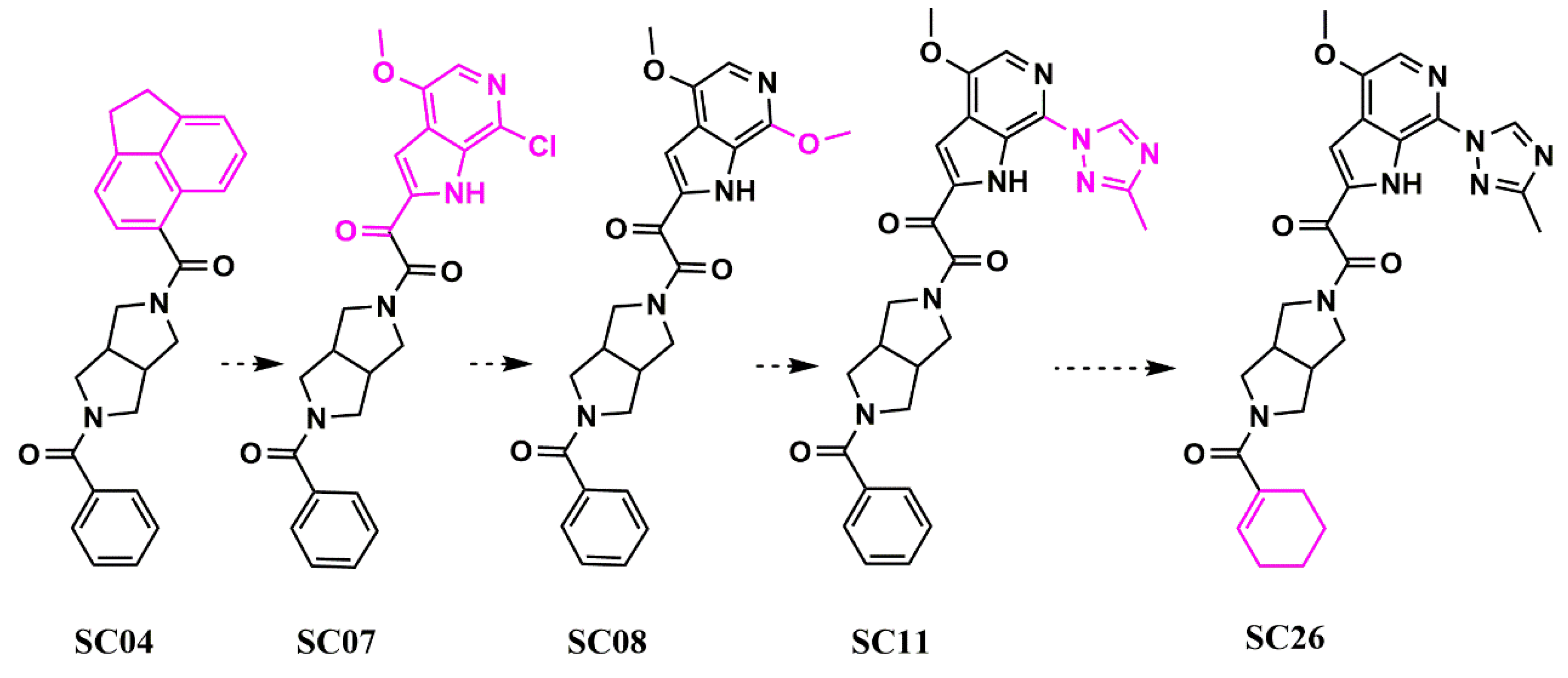
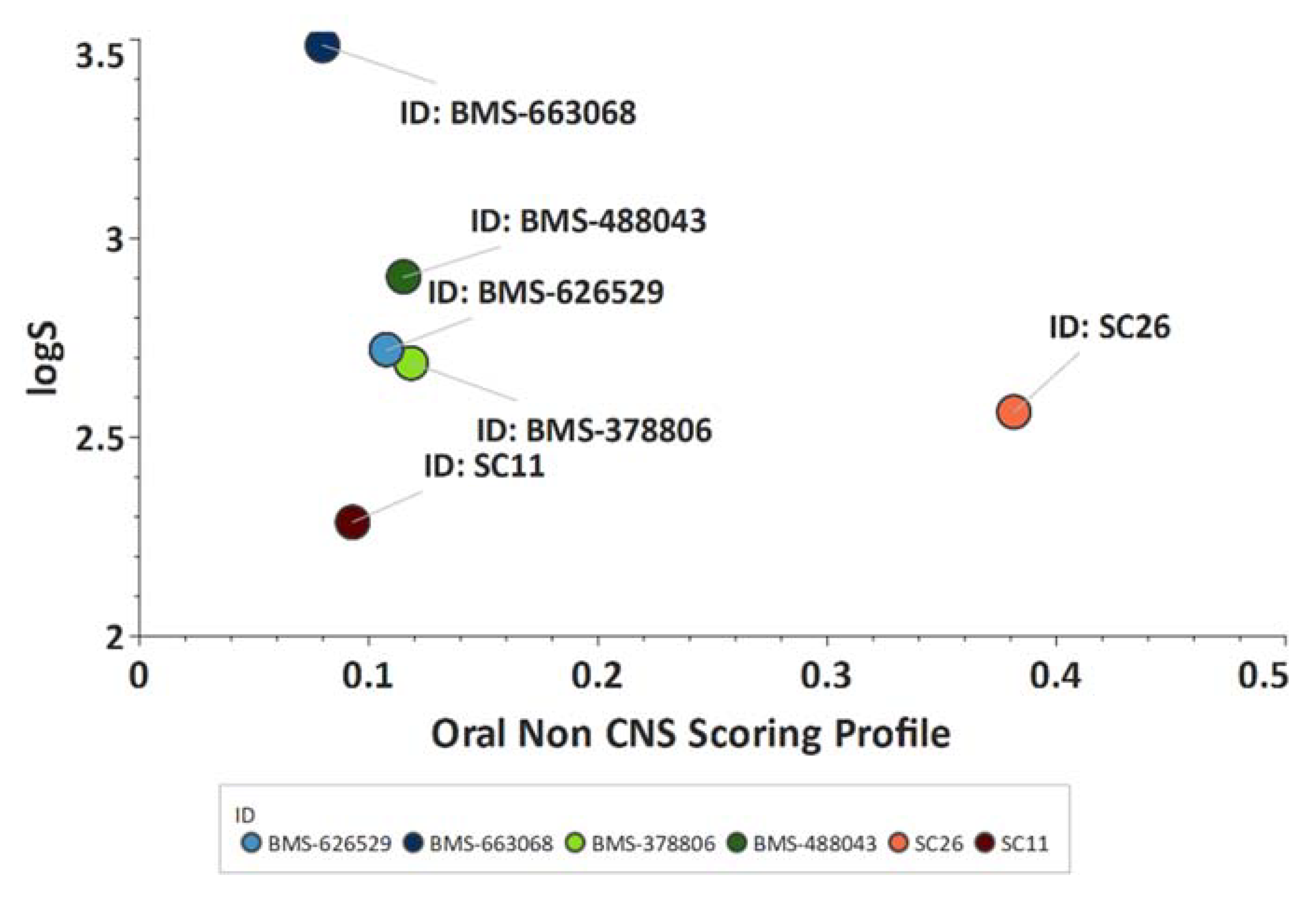
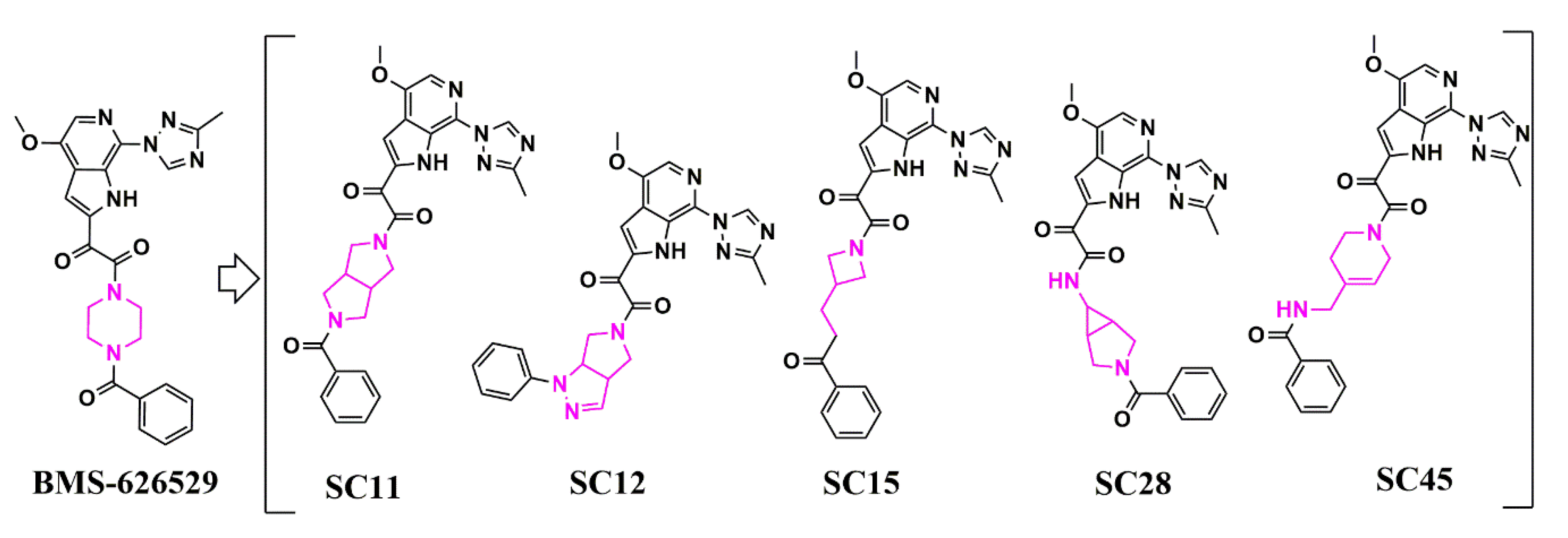
|
Compound |
IC50 JR-CSF |
IC50 B41 |
IC50 HxBc2 |
IC50 AMLV |
|---|---|---|---|---|
|
SC11 |
0.0008 ± 0.0001 |
0.002 ± 0.0002 |
0.001 ± 0.0001 |
N.A. |
|
SC12 |
0.008 ± 0.002 |
0.006 ± 0.003 |
0.080 ± 0.020 |
N.A. |
|
SC15 |
0.003 ± 0.001 |
0.007 ± 0.001 |
0.009 ± 0.001 |
N.A. |
|
SC28 |
0.096 ± 0.019 |
0.085 ± 0.03 |
0.069 ± 0.014 |
N.A. |
|
SC45 |
0.224 ± 0.017 |
0.350 ± 0.030 |
0.380 ± 0.030 |
N.A. |
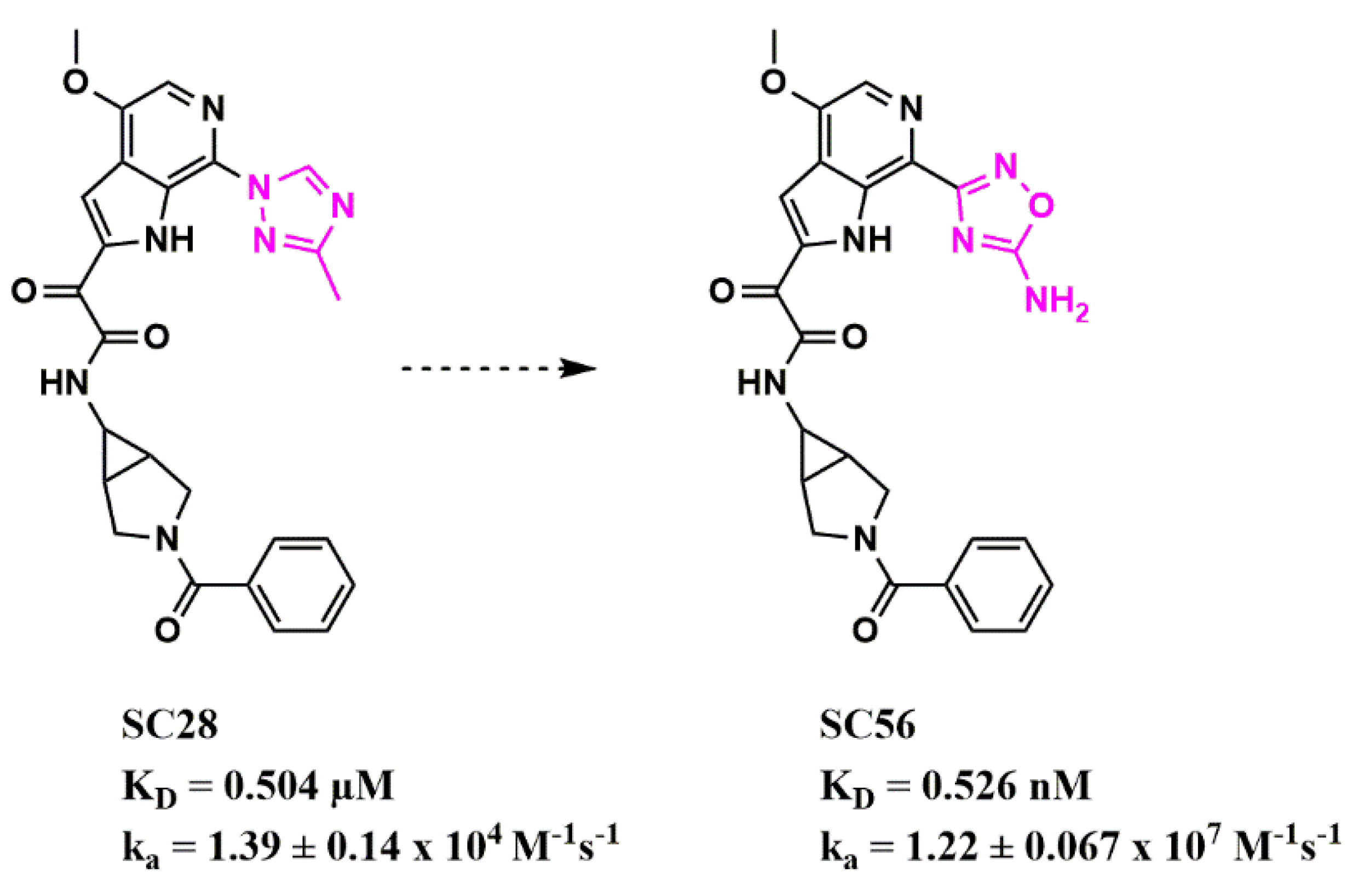
References
- Langmuir, I. Isomorphism, Isosterism and Covalence. J. Am. Chem. Soc. 1919, 41, 1543–1559.
- Grimm, H.G. Structure and Size of the Non-metallic Hydrides. Z. Electrochem. 1925, 31, 474–480.
- Grimm, H.G. On the Systematic Arrangement of Chemical Compounds from the Perspective of Research on Atomic Composition; and on Some Challenges in Experimental Chemistry. Naturwissenschaften 1929, 17, 557–564.
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176.
- Friedman, H.L. Influence of Isosteric Replacements upon Biological Activity. Nasnrs 1951, 206, 295–358.
- Burger, A. Isosterism and Bioisosterism in Drug Design. In Progress in Drug Research/Fortschritte der Arzneimittelforschung/Progrès des Recherches Pharmaceutiques; Birkhäuser: Basel, Switzerland, 1991.
- Doak, G.O.; Freedman, L.E. Medicinal Chemistry, 3rd ed.; Wiley-Interscience: New York, NY, USA, 1970.
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Modeling 2006, 46, 665–676.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field technology applied to virtual screening and finding the bioactive conformation. Expert Opin. Drug Discov. 2007, 2, 131–144.
- Cheeseright, T.J.; Holm, M.; Lehmann, F.; Luik, S.; Gottert, M.; Melville, J.L.; Laufer, S. Novel lead structures for p38 MAP kinase via FieldScreen virtual screening. J. Med. Chem. 2009, 52, 4200–4209.
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual screening using molecular fields. Application to the DUD data set. J. Chem. Inf. Modeling 2008, 48, 2108–2117.
- Cheeseright, T.J.; Mackey, M.D.; Scoffin, R.A. High content pharmacophores from molecular fields: A biologically relevant method for comparing and understanding ligands. Curr. Comput. Aided Drug Des. 2011, 7, 190–205.
- Apaya, R.P.; Lucchese, B.; Price, S.L.; Vinter, J.G. The matching of electrostatic extrema: A useful method in drug design? A study of phosphodiesterase III inhibitors. J. Comput. Aided Mol. Des. 1995, 9, 33–43.
- Vinter, J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J. Comput. Aided Mol. Des. 1994, 8, 653–668.
- Ketas, T.J.; Schader, S.M.; Zurita, J.; Teo, E.; Polonis, V.; Lu, M.; Klasse, P.J.; Moore, J.P. Entry inhibitor-based microbicides are active in vitro against HIV-1 isolates from multiple genetic subtypes. Virology 2007, 364, 431–440.
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239.
- Landry, I.; Zhu, L.; Tarif, M.A.; Hruska, M.; Sadler, B.M.; Pitsiu, M.; Joshi, S.; Hanna, G.J.; Lataillade, M.; Boulton, D.W.; et al. Model-Based Phase 3 Dose Selection for HIV-1 Attachment Inhibitor Prodrug BMS-663068 in HIV-1-Infected Patients: Population Pharmacokinetics/Pharmacodynamics of the Active Moiety, BMS-626529. Antimicrob. Agents Chemother. 2016, 60, 2782–2789.
- Nowicka-Sans, B.; Gong, Y.F.; McAuliffe, B.; Dicker, I.; Ho, H.T.; Zhou, N.; Eggers, B.; Lin, P.F.; Ray, N.; Wind-Rotolo, M.; et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507.
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorganic Med. Chem. Lett. 2014, 24, 5439–5445.
- Lin, P.F.; Blair, W.; Wang, T.; Spicer, T.; Guo, Q.; Zhou, N.; Gong, Y.F.; Wang, H.G.; Rose, R.; Yamanaka, G.; et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 2003, 100, 11013–11018.
- Yang, Z.; Zadjura, L.M.; Marino, A.M.; D’Arienzo, C.J.; Malinowski, J.; Gesenberg, C.; Lin, P.F.; Colonno, R.J.; Wang, T.; Kadow, J.F.; et al. Utilization of in vitro Caco-2 permeability and liver microsomal half-life screens in discovering BMS-488043, a novel HIV-1 attachment inhibitor with improved pharmacokinetic properties. J. Pharm. Sci. 2010, 99, 2135–2152.
- Tuyishime, M.; Lawrence, R.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorganic Med. Chem. Lett. 2016, 26, 228–234.
- Meuser, M.E.; Rashad, A.A.; Ozorowski, G.; Dick, A.; Ward, A.B.; Cocklin, S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules 2019, 24, 1581.

