Bioisosteric replacement is a powerful tool for modulating the drug-like properties, toxicity, and chemical space of experimental therapeutics. The use of bioisosteres and the introduction of structural changes to the lead compound allows the chemist to alter the compound’s size, shape, electronic distribution, polarizability, dipole, polarity, lipophilicity, and pKa, while still retaining potent target engagement.
1. Introduction
The design and development of a lead compound into a drug is a laborious and often costly process, with most candidates failing due to metabolism and pharmacokinetics issues rather than potency. Bioisosteric replacement is a strategy used by medicinal chemists to address these limitations while still retaining the potency/efficacy of the initial lead compound. The use of bioisosteres and the introduction of structural changes to the lead compound allows the chemist to alter the compound’s size, shape, electronic distribution, polarizability, dipole, polarity, lipophilicity, and pKa, while still retaining potent target engagement. Therefore, the bioisosteric approach can be used for the rational modification of a lead compound towards a more attractive therapeutic agent with improved potency, selectivity, altered physical, metabolic, toxicological properties with the bonus of generating novel intellectual property (IP).
2. Principle of Bioisosterism and Historical Background
The term isosterism was first introduced by Irving Langmuir in 1919 during his studies on similarities of physicochemical properties of atoms, groups and molecules
[1]. He described compounds or groups of atoms with the same number of atoms and electrons, such as N
2 and CO, N
2O and CO
2, or N
3- and NCO
- as isosteres, and, based on these similarities of the arrangement of electrons, he defined 21 groups of isosteres. H. G. Grimm further developed this definition in the early 1920s. This early hypothesis of bioisosterism describes the ability of certain chemical groups to mimic other chemical groups
[2,3][2][3]. Accordingly, the addition of a hydride to an atom gives to the resulting pseudoatom the properties of the atom with the next highest atomic number (
Table 1)
[4].
Table 1.
Grimm’s Hydride Displacement Law.
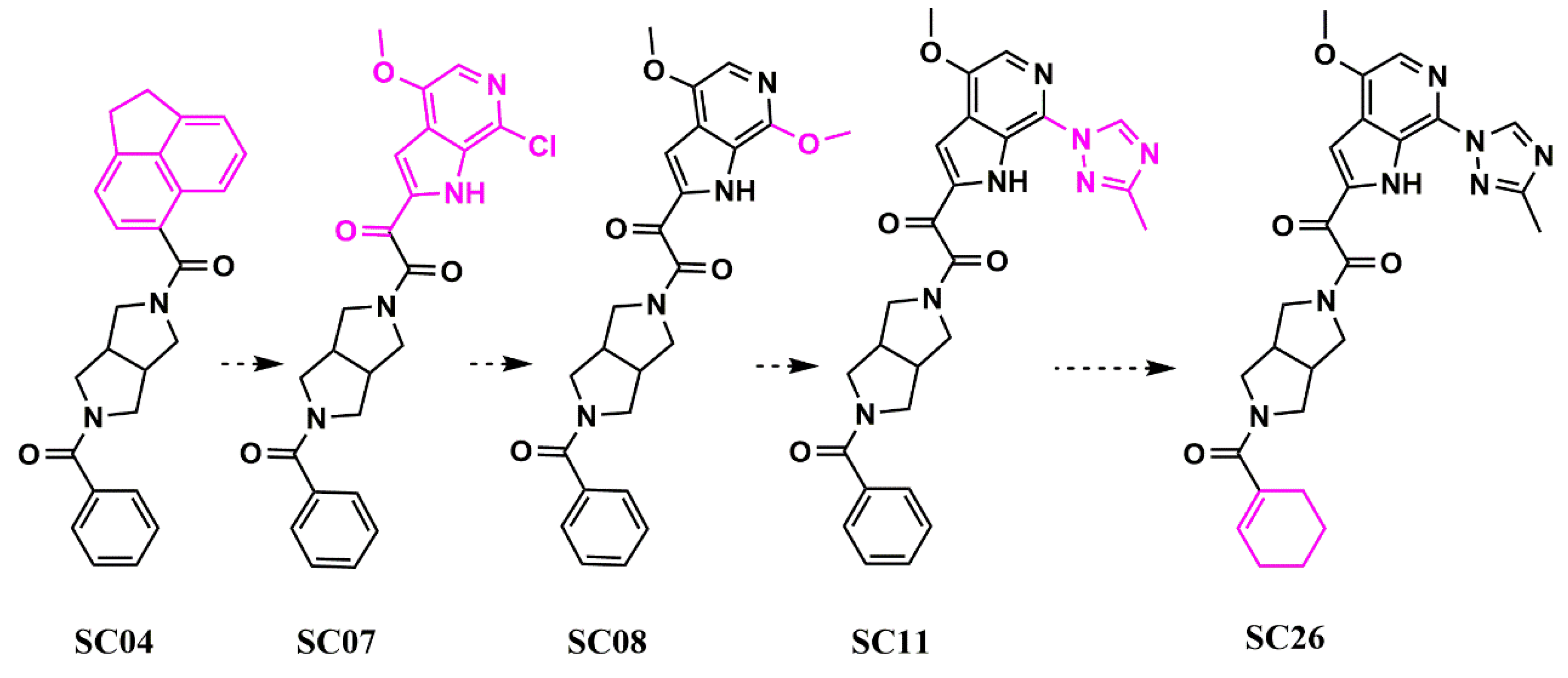
. Scaffold hopping in the core region of an HIV-1 entry inhibitor. Structural changes in the core region between each compound are highlighted in pink.
Table 3. Specificity and potency (in µM) of SC compounds against HIV-1J R-CSF, HIV-1 B41, HIV-1 HxBc2 Env, and HIV-1 AMLV pseudotyped HIV-1 using a single-round infection assay.
|
Compound
|
| F |
|
Ne
|
IC50 JR-CSF
|
IC50 B41
Na+
|
).
Table 2.
Isosteres based on valence electron number.
|
Number of Valence Electrons
|
|
| IC | 50 | HxBc2
|
IC50 AMLV
|
4
|
5
|
6
|
7
|
8
|
| |
CH
|
|
SC11
|
| NH
|
0.0008 ± 0.0001
OH
|
FH
|
|
| |
|
N+
|
P
|
S
|
0.002 ± 0.0002
Cl
|
| -
|
0.001 ± 0.0001 |
|
| ClH |
N.A.
|
|
|
|
P+
|
| CH |
As
2
|
Se
NH2
|
OH2
|
FH2+
|
|
SC12
|
0.008 ± 0.002
|
0.006 ± 0.003 |
Br
|
|
0.080 ± 0.020
BrH
|
N.A.
|
|
|
|
S+
|
| |
Sb
CH3
|
NH3
|
|
|
SC15
|
0.003 ± 0.001
|
Te
|
0.007 ± 0.001
| OH | 3+
|
|
| 0.009 ± 0.001 |
|
| N.A.
|
CH4
|
NH4+
|
Each vertical column represents an isostere, according to Grimm. In 1932, Hans Erlenmeyer extended Grimm’s definition of isosteres as atoms, ions, and molecules in which the peripheral layers of electrons (valence electrons) are considered as identical (
|
|
I |
|
|
|
| IH |
|
| |
|
As+ |
|
| |
|
SC28
| |
|
0.096 ± 0.019
PH
|
0.085 ± 0.03
SH
|
SH2
|
|
Sb+
|
|
|
PH2
|
PH3
|
Based on its application in biological systems, Harris Friedman introduced the term “bioisostere” in 1950 that included all atoms and molecules which fit the broadest definition for isosteres and have similar biological activity, either agonistic or antagonistic
[5]. Today, the even more broadened definition of bioisosteres introduced by Alfred Burger in the early 1990s is in use. Accordingly, bioisosteres are “Compounds or groups that possess near-equal molecular shapes and volumes, approximately the same distribution of electrons, and similar physical properties”
[6].
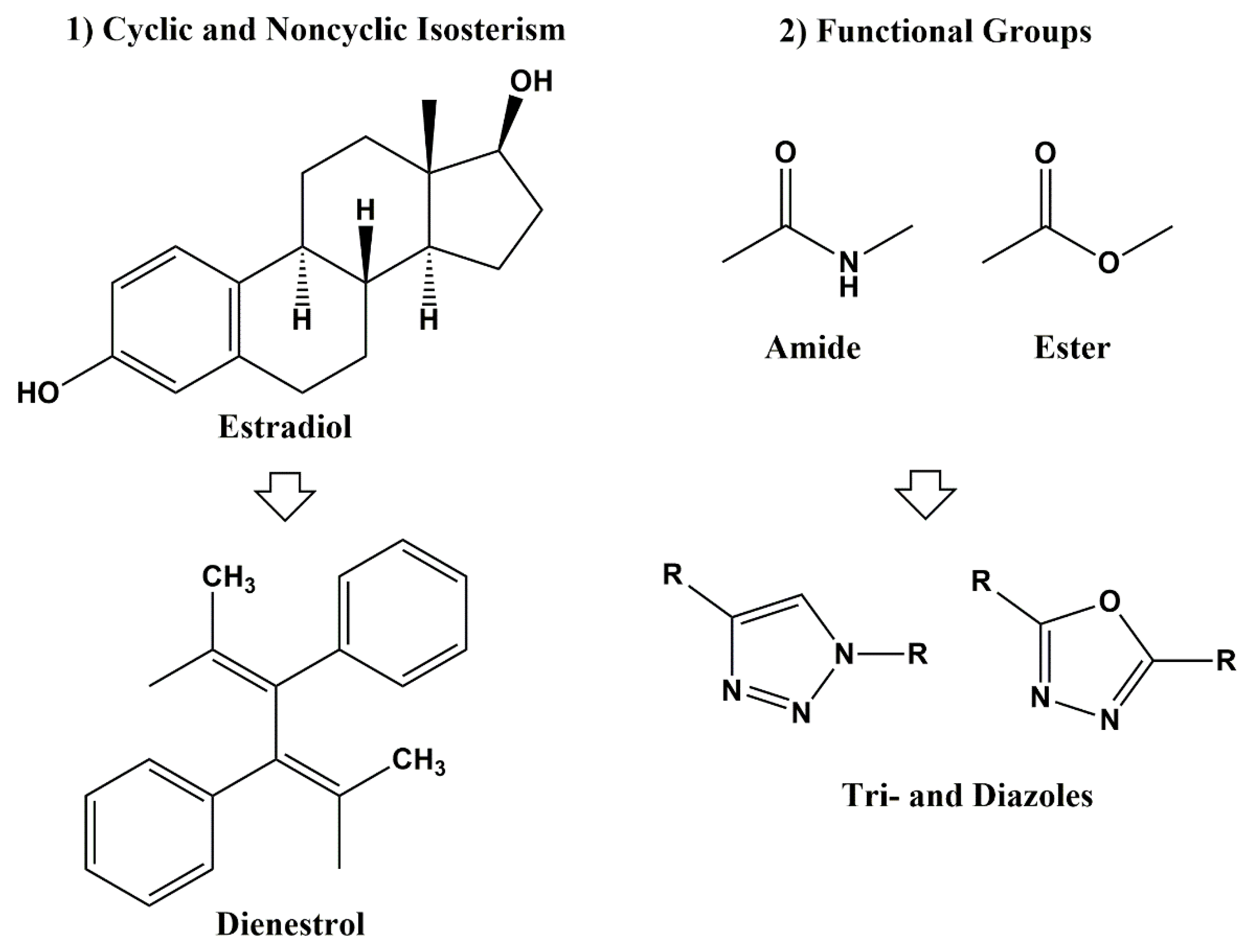
3. Classical and Non-Classical Bioisosteres
In the 1970s, Alfred Bruger defined bioisosteres as either classical (atom number, number of valence electrons, and degree of unsaturation) or non-classical (similar pKa, electrostatic potentials, orbital occupation/HOMOs and LUMOs)
[7]. Classical bioisosteres can be further subdivided into five classes: 1) monovalent atoms or groups (D and H; F and H; C and Si; Cl, Br, SH, and OH; NH and OH; RSH and ROH, –Cl, –PH
2, –SH), 2) divalent atoms or groups (–CH
2, –NH, –O, –S, –Se–, -COCH
2-), 3) trivalent atoms or groups (–CH=, –N=, -P=, -As=), 4) tetravalent atoms or groups (>C<, >Si< and =C=, =N
+=, =P
+=), and 5) ring equivalents (-CH=CH-, -S- (e.g., benzene, thiophene), -CH=, -N= (e.g., benzene, pyridine), -O-, -S-, -CH
2- (e.g., tetrahydrofuran, tetrahydrothiophene, cyclopentane)
[4,8][4][8]. Non-classical bioisosteres are a more sophisticated mimicry of the emulated counterparts and they do not strictly obey the steric and electronic definition of classical isosteres. As non-classical bioisosteres can significantly differ in electronic distribution, physicochemical, steric and topological properties, they have found beneficial applications in drug discovery research. Non-classical bioisosteres are subdivided into two groups: 1) cyclic vs. non-cyclic and 2) exchangeable functional groups (
Figure 1)
[4].
Figure 1.
Non-Classical Bioisosterism.
4. In Silico Molecular Field-Based Scaffold Hopping for Non-Classical Bioisostere Identification
Scaffold hopping using bioisosteric replacement has always been an integral part of the conventional drug development process but has expanded in recent years due to the availability of facile computational methods. High throughput screening (HTS), which tests thousands or millions of compounds, is a mainstay in the pharmaceutical industry. However, by applying computationally driven bioisosteric replacement to such initial hits, chemotype diversification can be readily achieved, which in turn increases the probability of successfully carrying one of these chemotypes through to the clinic.
Typical approaches that do not use molecular field points tend to view the bioisosteric fragment in a vacuum, taking only fragment comparison into account and not factoring in the effect of the replacement across the whole molecule. Using molecular fields to facilitate a comparison of the entire spatial physicochemical properties across the entire molecule, rather than the structure alone, can and has led to discoveries of non-intuitive bioisosteric replacements that have positively modulated drug-like properties while retaining target engagement
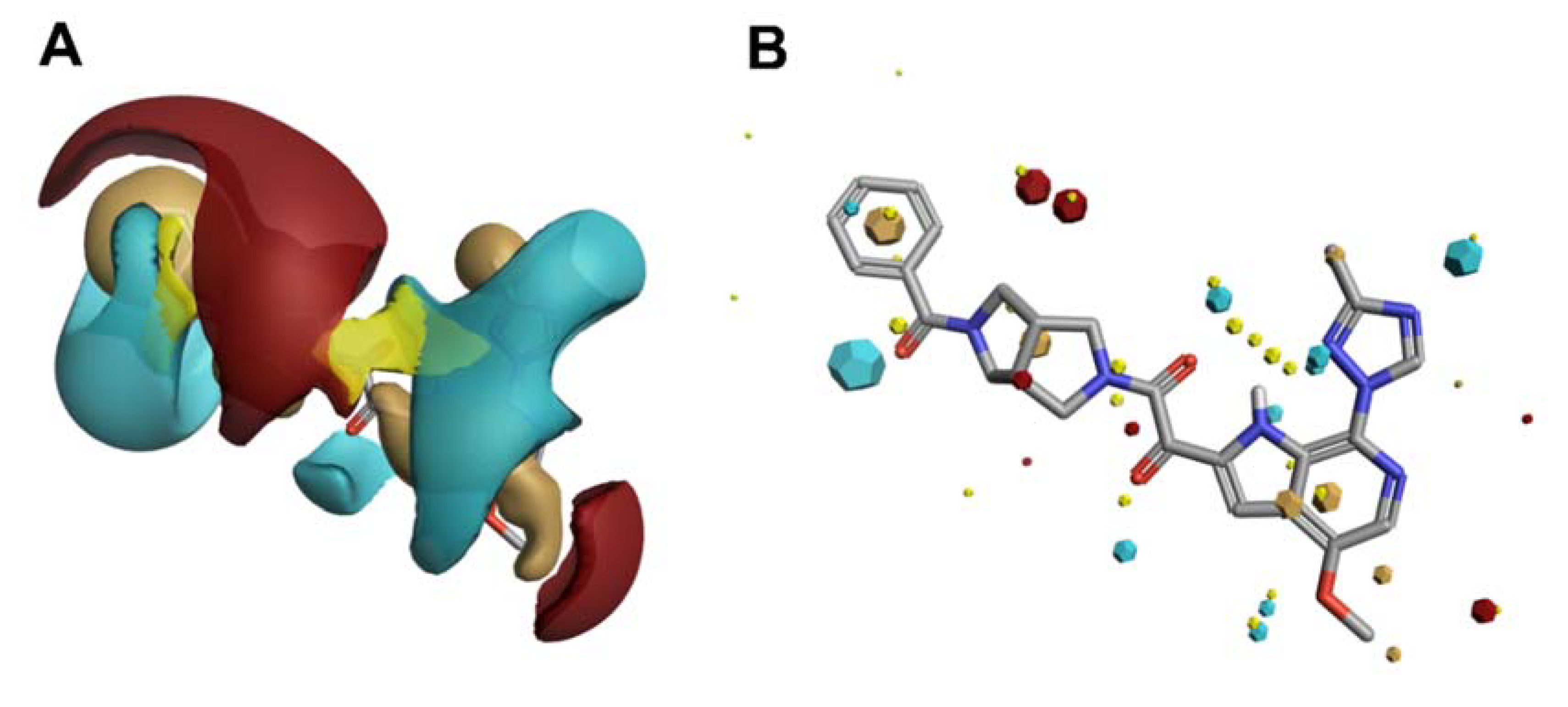
[39,40,41,42,43][9][10][11][12][13]. However, molecular fields are continuous and vary with the conformation of the compound, and sampling at defined points in the three-dimensional grid can be computationally expensive (
Figure 2A). Therefore, program collections such as Cresset (Cresset, Litlington, UK) use only extrema of these fields, the so-called field point, where intermolecular interactions most likely occur (
Figure 2B).
Figure 2. Electrostatic (blue/red), van der Waals (yellow) and hydrophobic (beige) contours (
A) and Field Point maxima (
B) of an HIV-1 entry inhibitor.
This calculation and comparison of field points is based on the eXtended Electron Density Force Field Theory (XED)
[44,45][14][15]. XED takes into account positive and negative electrostatic, Van der Waals, and hydrophobic field points. In contrast to most molecular mechanics force field methods that apply atom-centered partial charges, XED generates negative pseudo-orbitals surrounding the positive nuclei and distributes partial charges according to the orbital location. XED, therefore, mimics the electron distribution in a more accurate way by taking π-clouds and lone pairs into account, which play important roles in biological systems. In practice, Field Points of two or more compounds can be compared by sampling conformations to maximize the fields for optimal alignment, and in contrast to a structural alignment, new compounds with different structures but similar numbers and distribution of Field Points can be found. This so-called “templating” of molecules can also be used to generate a high-content pharmacophore for therapeutically interesting targets for which there is no known information regarding the small molecule binding site.
Scaffold Hopping for Potency and ADME Improvement of HIV-1 Entry Leads
The inhibition of HIV-1 entry is an attractive, yet underexploited therapeutic approach for several reasons. First, much of the entry process occurs in water-soluble compartments easily accessible to drugs. Second, both viral and host cell components involved in HIV-1 entry have been identified and can be targeted. Third, because inhibition of virus entry prevents the host cell from becoming permanently infected, such inhibitors are potentially useful in many different modalities, including pre-exposure prophylactics, prophylactics, and microbicides
[46][16].
In an effort to bring this class of inhibitor out of the realm of salvage therapy, we have applied both high-content, field-point pharmacophores to perform virtual screening, and iterative bioisosteric replacements, to identify novel leads for HIV-1 entry inhibitors. The most potent and broadly acting HIV-1 inhibitors to date are piperazine-based introduced by Bristol-Myers Squibb
[47][17]. However, this class has low bioavailability, resulting from low solubility and poor dissolution. Bristol-Myers Squibb has partially overcome these limitations by adopting a prodrug approach with BMS-663068 that shows increased solubility in the gut. However, after removal of the phosphonooxymethyl-solubilizing group in the gut, the properties of the active compound, BMS-626529, dominate and, despite its potency, 1.2 g per day is required to achieve an effective plasma concentration
[48][18]. Clearly, an entry inhibitor with more intrinsic drug-like properties would be preferable
[49][19]. Therefore, the researchers used field-based three-dimensional similarity virtual screening experiments using Blaze (Cresset, Litlington, UK) with a high-content field-based pharmacophore template derived from BMS-626529 and two of its predecessors, BMS-488043 and BMS-378806, to identify novel scaffolds that could function as entry inhibitors
[50][20]. Field templating was implemented,
as at the time of this study, no structural information regarding the binding site nor the bioactive conformation of the BMS compounds was available. FieldTemplater (Forge) allows the individual to generate a bioactive conformation hypothesis, and a field-point pattern pharmacophore, in the absence of structural information, as long as multiple, distinct compounds are known and are supposed to interact with the same site on the target.
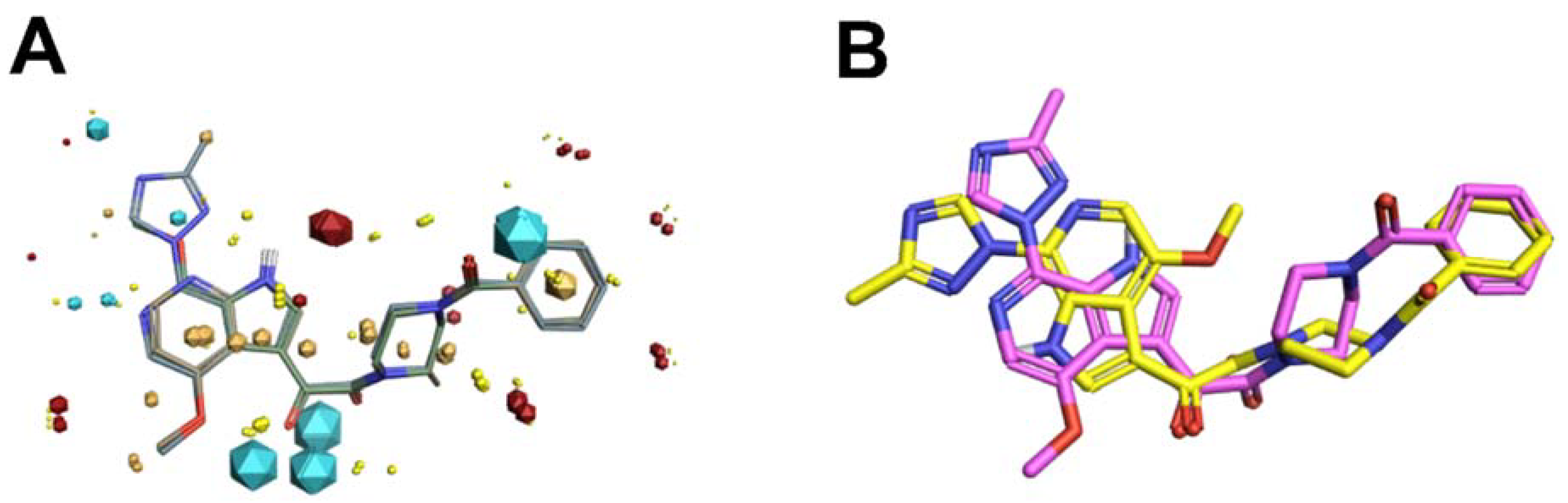
Figure 3 shows the field-point template generated and used in the reasearhers' HIV-1 entry inhibitor studies (
Figure 3A), in addition to a comparison of the template to the experimentally determined bioactive conformation of BMS-626529 (
Figure 3B)
[51,52][21][22].
Figure 3. (
A) Field-based template containing a single conformation of compounds BMS-378806 (pink), BMS-488043 (lime green), and BMS-626529 (teal green) aligned based on their three-dimensional field point patterns. Negatively charged field points are shown in blue; positively charged field points are red; van der Waals/shape field points are displayed in yellow; centers of hydrophobicity are shown in orange. The dodecahedral size of the field points is proportional to the magnitude of the extrema. (
B) Structural superimposition of BMS-62529 conformer generated with FieldTemplater (pink) and experimentally obtained crystal structure in complex with HIV-1 BG505 SOSIP.664 (yellow, PDB code: 5U7O).
From this field-point pharmacophore screen using Blaze (Cresset UK),
wthe
researchers identified hits with piperazine-based cores similar to the BMS chemotypes but with potencies in the low micromolar range, ranking from 13 to 153 µM using a HIV-1 YU-2 Env pseudotyped virus in a single-round infection assay (SRIA). Therefore, to obtain a truly novel core scaffold,
wethe researchers chose to conduct bioisosteric replacement (scaffold hopping) with Spark (Cresset, UK). The results of this indicated that replacing the piperazine group with a dipyrrolidine moiety would be viable. The resulting compound (
SC04) showed lower potency than the piperazine-based compounds (IC
50 HIV-1 YU-2 = 70 ± 6 μM; IC
50 HIV-1 JR-CSF = 100 ± 30 μM using a pseudotyped virus in an SRIA) but demonstrated specificity, making it perfect for further optimization in potency (
Figure 4).
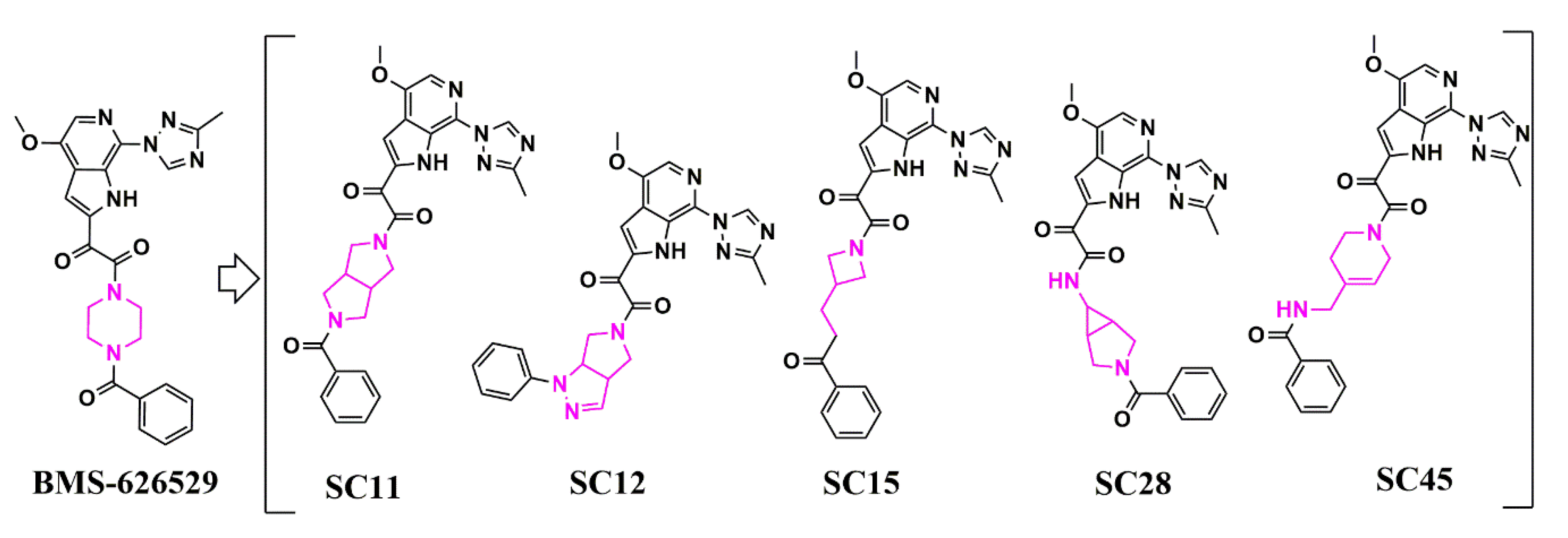
Figure 4. Evolution (from left to right) of HIV-1 entry inhibitors to improve potency and ADME properties. Structural changes between each compound are highlighted in pink.
Using Spark (Cresset, UK) and a fragment library generated from PubChem by fragmenting compounds with similarities to BMS-488043,
wthe researche
rs researched whether the dipyrrolidine could support nanomolar potency, whilst retaining specificity. Sequentially,
we the researchers generated
SC07 (IC
50 HIV-1 JR-CSF = 0.98 ± 0.06 μM in an SRIA),
SC08 (IC
50 HIV-1 JR-CSF = 0.09 ± 0.01 μM in an SRIA), and
SC11 (IC
50 HIV-1 JR-CSF values of 0.0008 ± 0.0004 μM in an SRIA). This was the first time that the piperazine core in this class of inhibitors had been successfully changed whilst retaining high potency. Finally, the replacement of the terminal phenyl in
SC11 by cyclohexene in
SC26 resulted in the first dipyrolrolodine-scaffolded highly potent HIV-1 entry inhibitor (IC
50 HIV-1 JR-CSF of 2.0 ± 0.1 nM; IC
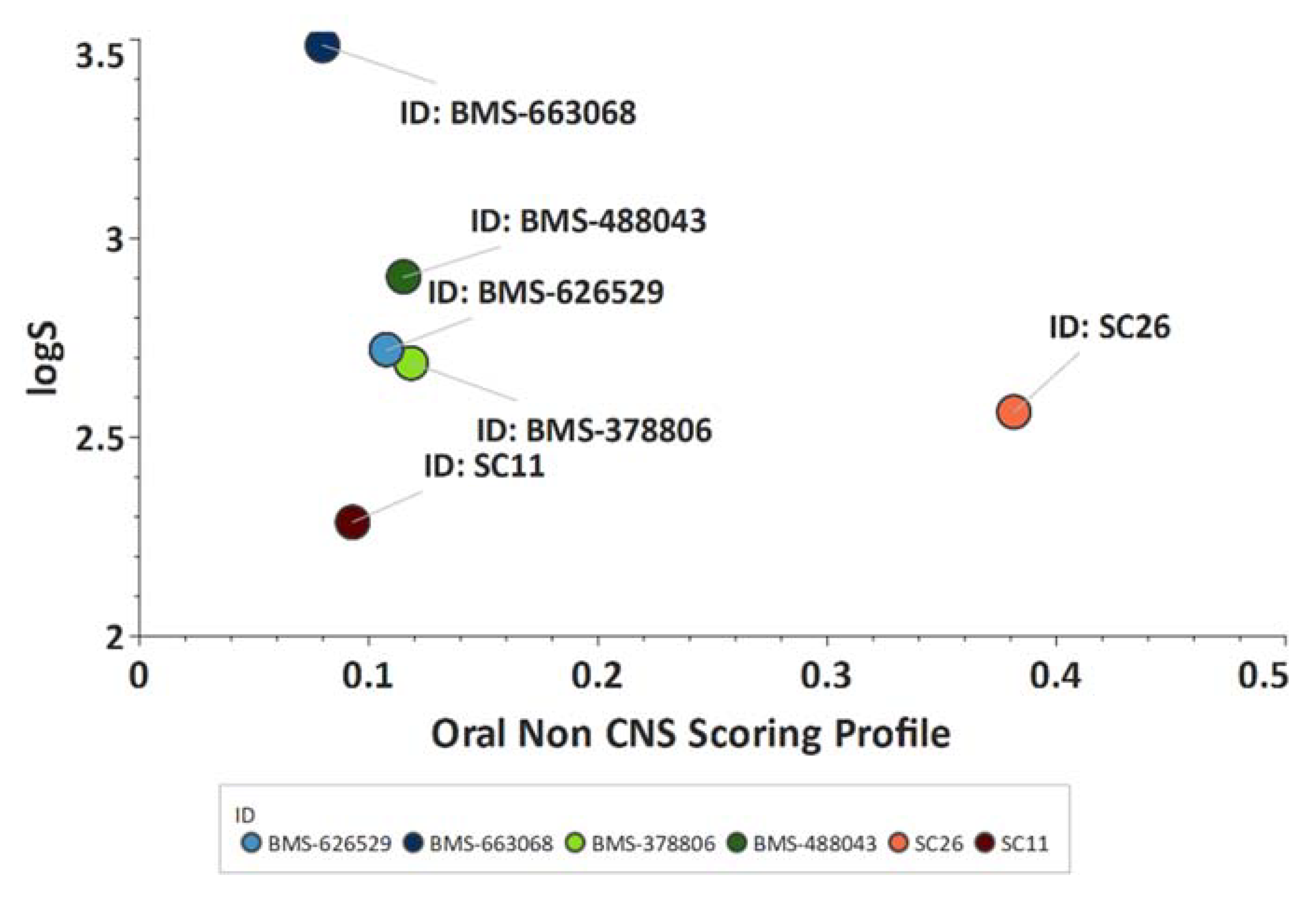
50 HIV-1 HxBc2 of 0.6 ± 0.01 nM in an SRIA), with significantly improved predicted ADME properties (as indicated by an increase in the Oral Non-CNS Drug-like Score in StarDrop, Optibrium, UK;
Figure 45).
Figure 45. Improved ADME properties of
SC26. Plot showing the StarDrop (Optibrium, Ltd., Cambridge, UK)–derived logS versus the score from a multimetric oral non-CNS profile for the BMS and SC compounds. Scores range from 0 to 1, with 0 suggesting extremely non-drug-like, and 1 suggesting the perfect drug.
Following this initial success,
wthe researche
rs then used similar methods to extend the core chemotypes in the HIV-1 entry inhibitor class. This resulted in the creation of five distinct core chemotypes from the original piperazine BMS-626529 that support specificity and nanomolar potencies (
Figure 56 and
Table 3)
[53][23].
Figure 56
|
|
0.069 ± 0.014 |
|
|
|
| N.A. |
|
|
SC45
|
0.224 ± 0.017
|
0.350 ± 0.030
|
0.380 ± 0.030
|
N.A.
|
N.A. = not active; N.D. = not determined.
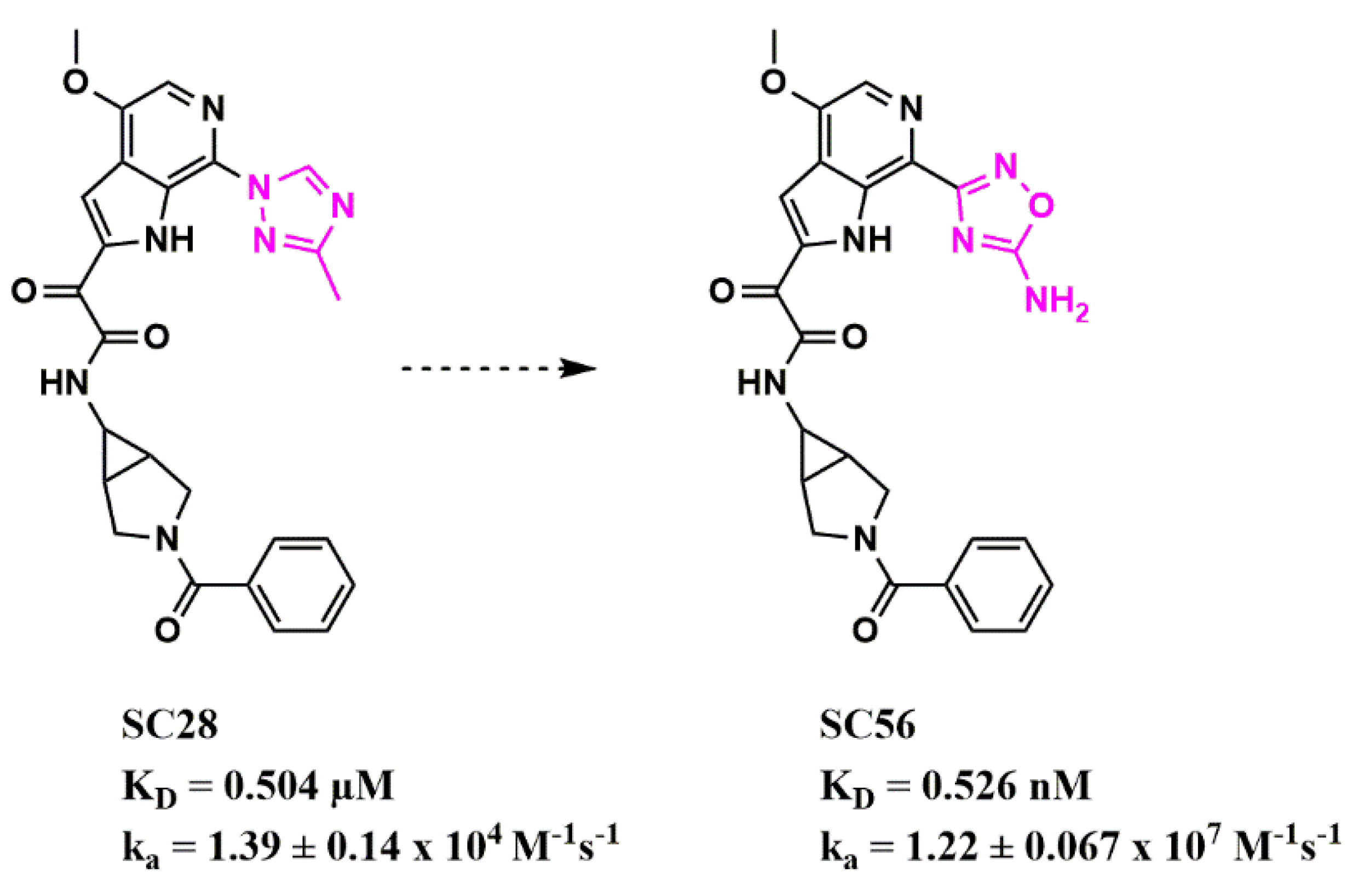
Stemming from these initial successes,
wthe researche
rs have since applied this workflow to identify new bioisosteres for the methyltriazole-azaindole moiety, as the main potency determinant of this entry inhibitor class and identified a compound (SC56), wherein the methyltriazole in SC28 was replaced with an amine-oxadiazole (
Figure 67)
[54][24]. This compound displayed a 1000-fold increased affinity for the B41 SOSIP Env, as judged by surface plasmon resonance (SPR).
Figure 67. Modulation of binding kinetics using bioisosteric replacement in HIV-1 entry inhibitor design. The structural change between the two compounds is highlighted in pink. Affinity and kinetic parameters were determined using a surface plasmon resonance assay.
Finally, in a recently published study,
wthe researche
rs have also demonstrated that SC28, with the azabicyclo-hexane core scaffold, has twice the metabolic stability of BMS-626529
[1]. This approach highlights bioisosteric replacement as a powerful tool that can be utilized to rapidly diversify chemotypes but can improve drug–target kinetic profiles and metabolism in the drug development process.