 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matthew K. Boag | + 2577 word(s) | 2577 | 2022-03-02 04:11:53 | | | |
| 2 | Rita Xu | Meta information modification | 2577 | 2022-03-03 02:42:19 | | | | |
| 3 | Rita Xu | + 28 word(s) | 2605 | 2022-03-11 11:11:00 | | |
Video Upload Options
A major hallmark of Parkinson’s disease (PD) is the fatal destruction of dopaminergic neurons within the substantia nigra pars compacta. This event is preceded by the formation of Lewy bodies, which are cytoplasmic inclusions composed of α-synuclein protein aggregates. A triad contribution of α-synuclein aggregation, iron accumulation, and mitochondrial dysfunction plague nigral neurons, yet the events underlying iron accumulation are poorly understood. Elevated intracellular iron concentrations up-regulate ferritin expression, an iron storage protein that provides cytoprotection against redox stress. The intrinsically disordered synaptic protein, α-synuclein, is the principal component of neuronal Lewy bodies (LB) and Lewy neurites (LN), which are cytoplasmic inclusions that hallmark α-synucleinopathies.
1. Introduction
2. Roles of Iron, Calcium and α-Synuclein in Nigral Neurons
3. Synaptic Role of α-Synuclein
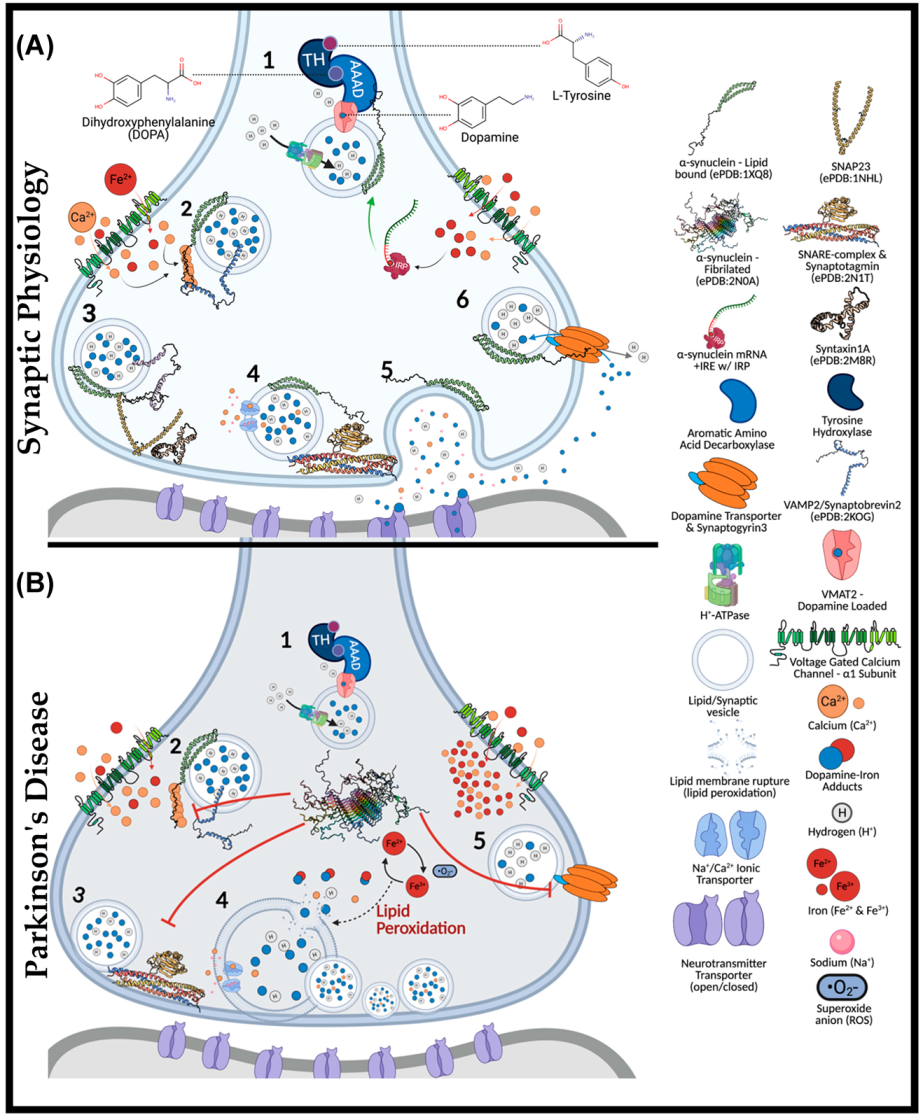
4. Iron and Calcium Regulation of α-Synuclein
4.1. Aggregation Induction of α-Synuclein
4.2. Iron-Induced Catecholamine Oxidation and Redox Damage
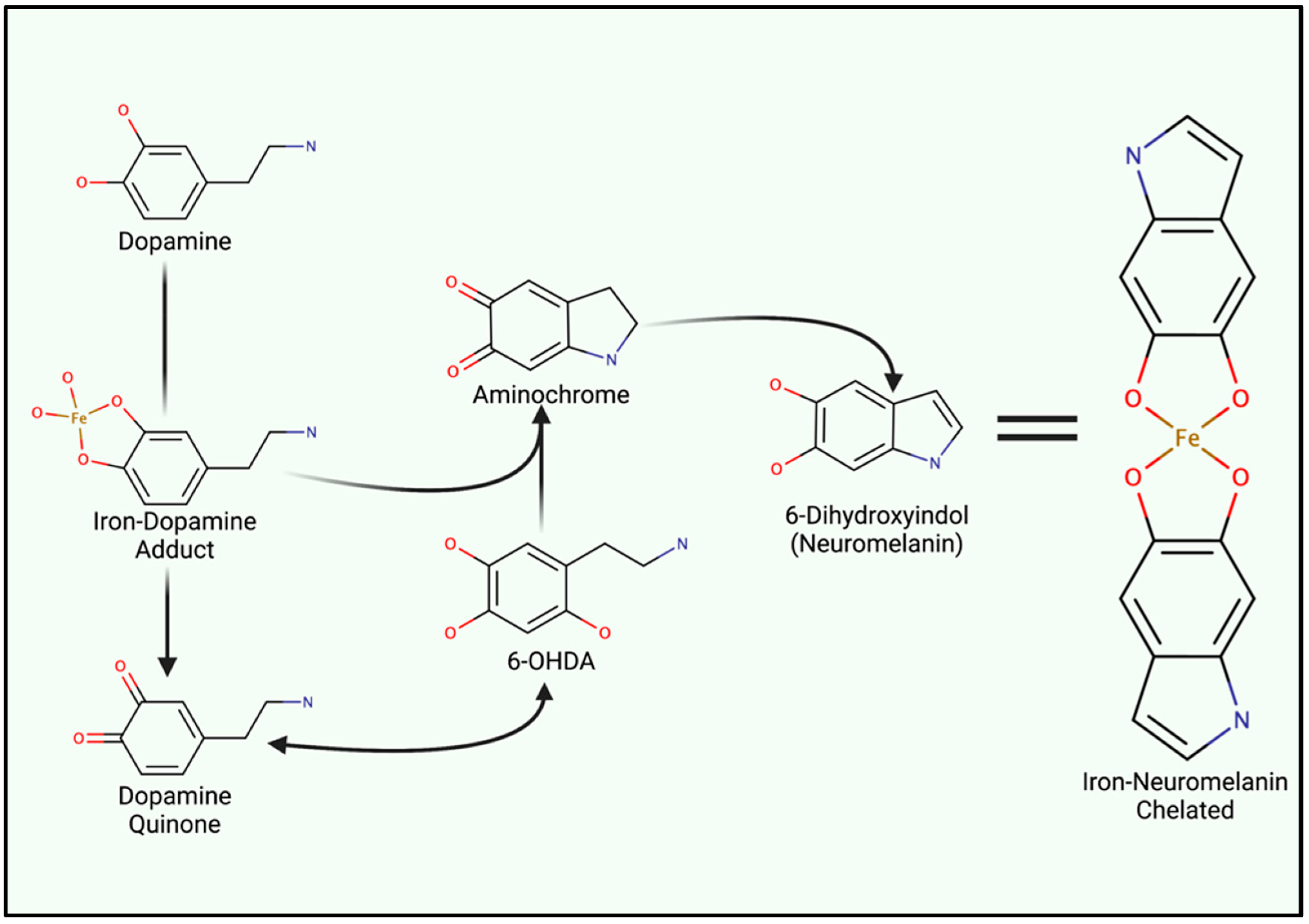
5. Iron Entry, Regulation and Cellular Metabolism
References
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236.
- Watanabe, I.; Vachal, E.; Tomita, T. Dense core vesicles around the Lewy body in incidental Parkinson’s disease: An electron microscopic study. Acta Neuropathol. 1977, 39, 173–175.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm 2020, 127, 181–187.
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264.
- Abeyawardhane, D.L.; Lucas, H.R. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxid. Med. Cell Longev. 2019, 2019, 4609702.
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975.
- Zecca, L.; Shima, T.; Stroppolo, A.; Goj, C.; Battiston, G.A.; Gerbasi, R.; Sarna, T.; Swartz, H.M. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience 1996, 73, 407–415.
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542.
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7.
- Calo, L.; Wegrzynowicz, M.; Santivanez-Perez, J.; Grazia Spillantini, M. Synaptic failure and alpha-synuclein. Mov. Disord. 2016, 31, 169–177.
- Di Marco Vieira, B.; Radford, R.A.W.; Hayashi, J.; Eaton, E.D.; Greenaway, B.; Jambas, M.; Petcu, E.B.; Chung, R.S.; Pountney, D.L. Extracellular Alpha-Synuclein Promotes a Neuroinhibitory Secretory Phenotype in Astrocytes. Life 2020, 10, 183.
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.M.; Sobott, F. Metal ions shape alpha-synuclein. Sci. Rep. 2020, 10, 16293.
- Han, J.; Day, J.R.; Connor, J.R.; Beard, J.L. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J. Nutr. 2002, 132, 2769–2774.
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 2009, 24, 673–684.
- Meyron-Holtz, E.G.; Cohen, L.A.; Fahoum, L.; Haimovich, Y.; Lifshitz, L.; Magid-Gold, I.; Stuemler, T.; Truman-Rosentsvit, M. Ferritin polarization and iron transport across monolayer epithelial barriers in mammals. Front. Pharmacol. 2014, 5, 194.
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352.
- Everett, J.; Brooks, J.; Lermyte, F.; O’Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. Iron stored in ferritin is chemically reduced in the presence of aggregating Abeta(1-42). Sci. Rep. 2020, 10, 10332.
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114.
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018, 233, 9179–9190.
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238.
- Hodge, G.K.; Butcher, L.L. Pars compacta of the substantia nigra modulates motor activity but is not involved importantly in regulating food and water intake. Naunyn. Schmiedebergs Arch. Pharmacol. 1980, 313, 51–67.
- Fabbri, M.; Reimao, S.; Carvalho, M.; Nunes, R.G.; Abreu, D.; Guedes, L.C.; Bouca, R.; Lobo, P.P.; Godinho, C.; Coelho, M.; et al. Substantia Nigra Neuromelanin as an Imaging Biomarker of Disease Progression in Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 491–501.
- Liang, C.L.; Sinton, C.M.; Sonsalla, P.K.; German, D.C. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 1996, 5, 313–318.
- de Berker, A.O.; Rutledge, R.B. A role for the human substantia nigra in reinforcement learning. J. Neurosci. 2014, 34, 12947–12949.
- Galtieri, D.J.; Estep, C.M.; Wokosin, D.L.; Traynelis, S.; Surmeier, D.J. Pedunculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife 2017, 6, e30352.
- Sonne, J.; Reddy, V.; Beato, M.R. Neuroanatomy, Substantia Nigra. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152.
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829.
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926.
- Melland, H.; Carr, E.M.; Gordon, S.L. Disorders of synaptic vesicle fusion machinery. J. Neurochem. 2021, 157, 130–164.
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; O’Donnell, P.; Drobatz, K.; Gurda, B.; et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J. Pharmacol. Exp. Ther. 2016, 358, 254–261.
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thevenod, F. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145.
- Du, X.; Xu, H.; Shi, L.; Jiang, Z.; Song, N.; Jiang, H.; Xie, J. Activation of ATP-sensitive potassium channels enhances DMT1-mediated iron uptake in SK-N-SH cells in vitro. Sci. Rep. 2016, 6, 33674.
- Bazelon, M.; Fenichel, G.M.; Randall, J. Studies on neuromelanin. I. A melanin system in the human adult brainstem. Neurology 1967, 17, 512–519.
- Carballo-Carbajal, I.; Laguna, A.; Romero-Gimenez, J.; Cuadros, T.; Bove, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Penuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973.
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712.
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Trexler, A.J.; Rhoades, E. N-Terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 2012, 21, 601–605.
- Deng, S.; Pan, B.; Gottlieb, L.; Petersson, E.J.; Marmorstein, R. Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. Elife 2020, 9, e57491.
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124.
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449.
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475.
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173.
- Hong, W.; Lev, S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35–43.
- Hawk, B.J.D.; Khounlo, R.; Shin, Y.K. Alpha-Synuclein Continues to Enhance SNARE-Dependent Vesicle Docking at Exorbitant Concentrations. Front. Neurosci. 2019, 13, 216.
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115.
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Sudhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122.
- Koo, S.J.; Markovic, S.; Puchkov, D.; Mahrenholz, C.C.; Beceren-Braun, F.; Maritzen, T.; Dernedde, J.; Volkmer, R.; Oschkinat, H.; Haucke, V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. USA 2011, 108, 13540–13545.
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283.
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159.
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39.
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396.
- Sharma, M.; Burre, J.; Bronk, P.; Zhang, Y.; Xu, W.; Sudhof, T.C. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012, 31, 829–841.
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763.
- Zhou, P.; Pang, Z.P.; Yang, X.; Zhang, Y.; Rosenmund, C.; Bacaj, T.; Sudhof, T.C. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. 2013, 32, 159–171.
- Wang, S.; Li, Y.; Gong, J.; Ye, S.; Yang, X.; Zhang, R.; Ma, C. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat. Commun. 2019, 10, 69.
- Shao, K.; Li, F.; Yang, Y.; Wang, N.; Gao, X.D.; Nakanishi, H. Characteristics of SNARE proteins are defined by distinctive properties of SNARE motifs. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129658.
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Gotz, J.; Parton, R.G.; Collins, B.M.; et al. Munc18-1 is a molecular chaperone for alpha-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 2016, 214, 705–718.
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479.
- Lee, H.K.; Yang, Y.; Su, Z.; Hyeon, C.; Lee, T.S.; Lee, H.W.; Kweon, D.H.; Shin, Y.K.; Yoon, T.Y. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 2010, 328, 760–763.
- Zhou, Q.; Zhou, P.; Wang, A.L.; Wu, D.; Zhao, M.; Sudhof, T.C.; Brunger, A.T. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548, 420–425.
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252.
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578.
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554.
- Ishibashi, K.; Oda, K.; Ishiwata, K.; Ishii, K. Comparison of dopamine transporter decline in a patient with Parkinson’s disease and normal aging effect. J. Neurol. Sci. 2014, 339, 207–209.
- Egana, L.A.; Cuevas, R.A.; Baust, T.B.; Parra, L.A.; Leak, R.K.; Hochendoner, S.; Pena, K.; Quiroz, M.; Hong, W.C.; Dorostkar, M.M.; et al. Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-.-3. J. Neurosci. 2009, 29, 4592–4604.
- Cooper, A.A. Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328.
- Baksi, S.; Singh, N. alpha-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 2017, 7, 12843.
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and alpha-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 237.
- Llinas, R.; Sugimori, M.; Silver, R.B. Microdomains of high calcium concentration in a presynaptic terminal. Science 1992, 256, 677–679.
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, copper and alpha-Synuclein in alpha-synucleinopathies. Front. Neurosci. 2017, 11, 114.
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814.
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901.
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and alpha-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260.
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca(2+) Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004.
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902.
- Pountney, D.L.; Voelcker, N.H.; Gai, W.P. Annular alpha-synuclein oligomers are potentially toxic agents in alpha-synucleinopathy. Hypothesis. Neurotox. Res. 2005, 7, 59–67.
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar alpha-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513.
- Hindeya Gebreyesus, H.; Gebrehiwot Gebremichael, T. The Potential Role of Astrocytes in Parkinson’s Disease (PD). Med. Sci. 2020, 8, 7.
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5’-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 2007, 12, 222–223.
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896.
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035.
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119.
- Shima, T.; Sarna, T.; Swartz, H.M.; Stroppolo, A.; Gerbasi, R.; Zecca, L. Binding of iron to neuromelanin of human substantia nigra and synthetic melanin: An electron paramagnetic resonance spectroscopy study. Free Radic. Biol. Med. 1997, 23, 110–119.
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell Neurosci. 2020, 14, 218.
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848.
- Sulzer, D.; Cassidy, C.; Horga, G.; Kang, U.J.; Fahn, S.; Casella, L.; Pezzoli, G.; Langley, J.; Hu, X.P.; Zucca, F.A.; et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 11.
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030.
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614.
- del Toro, D.; Alberch, J.; Lazaro-Dieguez, F.; Martin-Ibanez, R.; Xifro, X.; Egea, G.; Canals, J.M. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 2009, 20, 1478–1492.
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590.
- Cho, S.J.; Yun, S.M.; Jo, C.; Lee, D.H.; Choi, K.J.; Song, J.C.; Park, S.I.; Kim, Y.J.; Koh, Y.H. SUMO1 promotes Abeta production via the modulation of autophagy. Autophagy 2015, 11, 100–112.
- Tadokoro, K.; Yamashita, T.; Shang, J.; Ohta, Y.; Nomura, E.; Morihara, R.; Omote, Y.; Takemoto, M.; Abe, K. Switching the Proteolytic System from the Ubiquitin-Proteasome System to Autophagy in the Spinal Cord of an Amyotrophic Lateral Sclerosis Mouse Model. Neuroscience 2021, 466, 47–57.
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281.
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339.
- Wilson, R.L.; Wightman, R.M. Systemic and Nigral Application of Amphetamine Both Cause an Increase in Extracellular Concentration of Ascorbate in the Caudate-Nucleus of the Rat. Brain Res. 1985, 339, 219–226.
- Schenk, J.O.; Miller, E.; Gaddis, R.; Adams, R.N. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982, 253, 353–356.
- Boag, M.K.; Ma, L.; Mellick, G.D.; Pountney, D.L.; Feng, Y.; Quinn, R.J.; Liew, A.W.-C.; Dharmasivam, M.; Azad, M.G.; Afroz, R.; et al. Calcium channels and iron metabolism: A redox catastrophe in Parkinson’s disease and an innovative path to novel therapies? Redox Biol. 2021, 47, 102136.

