Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
A major hallmark of Parkinson’s disease (PD) is the fatal destruction of dopaminergic neurons within the substantia nigra pars compacta. This event is preceded by the formation of Lewy bodies, which are cytoplasmic inclusions composed of α-synuclein protein aggregates. A triad contribution of α-synuclein aggregation, iron accumulation, and mitochondrial dysfunction plague nigral neurons, yet the events underlying iron accumulation are poorly understood. Elevated intracellular iron concentrations up-regulate ferritin expression, an iron storage protein that provides cytoprotection against redox stress.
- α-synuclein
- ferritin
- iron
- autophagy
- ferritinophagy
- vesicular trafficking
1. Introduction
Parkinson’s disease (PD) is the most prevalent degenerative motor disorder, characterized by dysfunction and death of dopaminergic neurons within the substantia nigra pars compacta (SNpc) [1][2][3][4][5]. The intrinsically disordered synaptic protein, α-synuclein, is the principal component of neuronal Lewy bodies (LB) and Lewy neurites (LN), which are cytoplasmic inclusions that hallmark α-synucleinopathies. Protein aggregation, mitochondrial dysfunction, and intracellular iron accumulation converge in a triad pathology [3][6][7][8] that progressively disperses across the neocortex [9][10][11][12].
Both iron and α-synuclein promote mitochondrial dysfunction, yet the aetiology of iron deregulation remains poorly understood [13]. Upon increased iron concentration within cells, up to 4500 Fe3+ ions per unit can be stored within ferritin, a universal intracellular iron storage protein [14][15][16][17][18]. Ferritinophagy is a subtype of the autophagy-lysosomal pathway and is the only known mechanism by which iron bound to ferritin can be released [19][20][21]. However, this degradative pathway depends on functional vesicular trafficking and membrane fusion events, which become inhibited by α-synuclein aggregates. Strong evidence suggests that dysfunctional ferritinophagy can potentiate iron overload in nigral neurons [21].
2. Roles of Iron, Calcium and α-Synuclein in Nigral Neurons
The SNpc is the most densely populated dopaminergic region, comprising 200,000 to 420,000 dopaminergic neurons, much of which die in PD patient tissue [8][22][23][24], ultimately manifesting into the cardinal symptoms of ataxia, bradykinesia, resting tremor, and a shuffling gait. Non-motor symptoms, such as cognitive dysfunction and decline, often occur at later disease stages but may also precede motor signs. In addition, both motor and cognitive dysfunction may be related to SNpc degeneration, as the cortical region is a crucial indirect regulator of voluntary motor control and behavioural learning [25].
Sensory stimuli transduction originates from the pedunculopontine nucleus (PPN), which transverses through the reticular activating system and then into the SNpc [26][27]. From here, a direct pathway exists (via dopamine D1 receptor stimulation) to propagate an excitatory stimulus for the overlying GABAergic (i.e., depending on γ-aminobutyric acid (GABA) substantia nigra pars reticulata (SNpr) [28]. GABAergic neurotransmission suppresses the internal globus pallidus, freeing the basolateral nuclei to stimulate the motor cortex. A parallel indirect pathway (via dopamine D2 receptors) excites GABAergic projections from the dorsal striatum. Subsequent repression of the external globus pallidus means the excitatory glutaminergic (i.e., glutamic acid neurotransmission) subthalamic nuclei can stimulate the internal globus pallidus. GABAergic signalling directed towards the thalamic basolateral nuclei completes an inhibitory circuit that regulates voluntary motor control, comprising the cortico-basal ganglia-thalamocortical loop [26]. This transduction circuit is implicated in numerous diseases, including PD, Huntington’s disease (HD), and neuropsychiatric disorders, such as attention-deficit hyperactivity disorder [29][30][31].
The SNpc is internally compartmentalized by calbindin-D28K (CALB1) expression, with an immuno-positive dorsal and negative ventral area [24][32]. In addition to selective expression of CALB1, the SNpc exhibits slow, broad oscillations that persistently supply the striatum with dopaminergic neurotransmission. Autonomous conduction stems from sporadic depolarization events that alter voltage-gated ion channels, dynamically fluctuating membrane potentials [33][34]. CALB1 probably combines with other physiological neuroprotectants (e.g., neuromelanin and metallothionein) to shield against the influx of detrimental ions. Neuromelanin (5,6-dihydroxyindole) is the darkly pigmented substance that has historically identified the SNpc [35] and is a chelator of metals and binds other reactive species [23][36]. PD histology exhibits the complete loss of SNpc pigmentation, indicating the death of all nigral neuromelanin-positive neurons [35].
3. Synaptic Role of α-Synuclein
Synaptic physiology and plasticity are modulated by α-synuclein (Figure 1). Upon correct folding of α-synuclein, the lipophilic N-terminus folds into twin helices that embed into small phospholipid vesicles (SMV) [37][38] and is supported by the N-acetylmethionine modification at residue M1 [39][40]. Despite being an intrinsically disordered cytosolic protein, the N-terminus of α-synuclein is highly conserved, with imperfect KTKEGV repeats that promote helix formation in the lipid-bound conformation [41]. The first helix extends from residues 1–25, while the parallel strand is between residues 31–55. Aspartate (D2) and glutamate (E13 and E20) are charged amino acids that stabilize the outer-surface residues, enhancing the lipid-binding of phosphoglycerol heads at lysine residues, K6, K10, and K12, at membrane surfaces [42]. Familial mutations within the SNCA gene (PARK1), including A30P, E46K, H50Q, G51D, A53E, and A53T, with sporadic PD-associated mutants, A18T and A29S, are also being linked to PD [43][44][45][46][47][48]. These α-synuclein mutants have diminished lipid binding, propagating disequilibrium towards non-membrane-bound monomers [43][44][45][46][47][48].
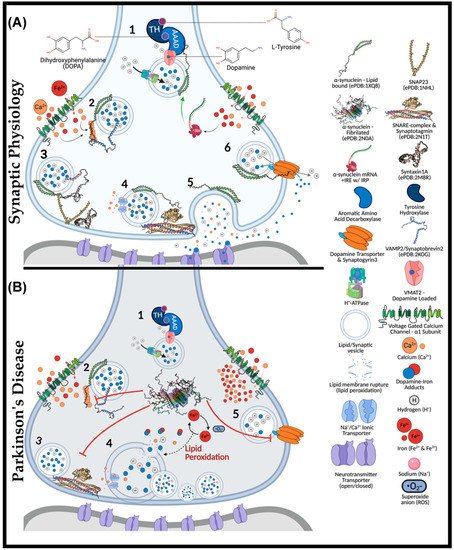
Figure 1. The Role of α-Synuclein at the Synaptic Junction in Physiology and in Parkinson’s Disease. In normal physiology (A): (1) complexed tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase (AAAD) bind to the vesicular monoamine transporter (VMAT2), yielding vesicular dopamine. Vesicle membrane-bound H+-ATPases lower the intra-vesicular pH. Lipid-vesicle embedded α-synuclein has a role (2) in chaperoning soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) after calcium activation. C-terminal binding between calcium-activated α-synuclein and vesicular-associated membrane protein 2 (VAMP2), exposes the SNARE motif for synaptosome-associated protein of 25 kDa (SNAP25) binding. SNAP25 recruits syntaxin-1A, and with the assistance of accessory proteins (synaptotagmin and Munc-18/13, an acronym for mammalian uncoordinated-18 or -13), the SNARE complex is formed (4), prompting the exocytosis of vesicular cargo (5). The α-synuclein is critical for synaptic plasticity by recycling synaptic vesicles post-exocytosis. Afterwards, (6) α-synuclein associates with the dopamine transporter to bring vesicles into proximity for synaptogyrin3 to modulate direct dopamine influx from the synaptic cleft. (B) (1) complexed tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase (AAAD) bind to the vesicular monoamine transporter (VMAT2), yielding vesicular dopamine. Iron has also been suggested to permeate voltage-gated calcium channels (VGCCs), increasing the intracellular iron pool that may drive α-synuclein synthesis via a controversial and atypical 5′ iron-responsive element (IRE) in the untranslated region of α-synuclein mRNA. However, in the absence of lipid membrane vesicles, excessive iron-induced α-synuclein synthesis can yield disordered species that readily fibrillate (2) upon calcium exposure. The α-synuclein aggregates bind VAMP2 C-termini to block SNARE complex assembly (3). Consequently, vesicles cluster at the presynaptic membrane (4), while iron redox chemistry can lead to lipid peroxidation. Integral membrane damage can rupture vesicles, expelling reactive catecholamines, which lead to further damage through redox-active dopamine-iron adducts. Dopamine recycling is halted (5) as α-synuclein aggregates block vesicles docking at the dopamine transporter (DAT).
Physiologically, α-synuclein is a calcium-sensing chaperone of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein complexes [49]. Calcium-binding enhances the hydrophobicity of α-synuclein for interactions with the vesicular-SNARE protein (v-SNARE or R-SNARE motif), including the vesicle-associated membrane protein 2 (VAMP2, also known as synaptobrevin) [50][51]. Synaptic exocytosis relies upon VAMP2, as demonstrated through knockout mice lacking evoked synaptic potential, thus being embryonically lethal [52][53]. VAMP2 mutations in the SNARE motif (A67P, S75P, F77S, & E77A) manifest severe neurodevelopmental disorders with cognitive deficits [54]. Interactions between calcium-bound α-synuclein (C-terminus) and VAMP2 (N-terminus) induce a structural reorganization of the latter, exposing the internal SNARE motif of VAMP2. If not already docked at presynaptic membranes, these events may parallel the Hsc70-CSPα-SGT chaperone complex [53][55][56].
Heat shock cognate 70 kDa (Hsc70) is an ATPase that covalently conjugates cysteine string protein-α (CSPα) to vesicular membranes for synaptic trafficking [56]. CSPα is another SNARE complex co-chaperone that also recruits small glutamate-rich tetratricopeptide repeat-containing protein (SGT). CSPα mechanistically serves to prime the target-SNARE (t-SNARE or Qb-SNARE-motif) and synaptosomal-associated protein of 25 kDa (SNAP25) at presynaptic membranes [57]. Interestingly, CSPα-knockout in mice causes significant neurodegeneration [58] that is rescued upon overexpressing human α-synuclein [57]. This result indicates that α-synuclein may analogously act to prime and stabilize SNAP25 in a compensatory manner. Another noteworthy point is that Hsc70 can directly bind fibrillated α-synuclein to facilitate chaperone-mediated autophagy (CMA) [59].
Unlike other SNARE proteins, SNAP25 does not contain a transmembrane domain and instead attaches to presynaptic membranes by palmitoylation at central cysteine residues [60]. The C- and N-termini of SNAP25 each contain a separate SNARE motif linking the proximal syntaxin-1a (Qa-SNARE motif) and vesicle-bound VAMP2 [61]. In attempts to prevent sporadic synaptic firing, syntaxin-1 remains independently regulated by mammalian uncoordinated 18 (Munc-18) and Munc-13 [62]. Munc-18 halts exocytosis by altering the conformation of syntaxin-1a. The Habc domain of synatxin-1a is rearranged to block exogenous interactions with the protein’s SNARE motif. Upon stimulation, Munc-13 switches with Munc-18 through their MUN domain (residues 29–96), ultimately releasing the folded syntaxin-1 for SNAP25 interactions [61][63]. It is unclear whether VAMP2 or α-synuclein may facilitate Munc-18/13 interchange. At this point, α-synuclein remains free of any known binding partners, whereas VAMP2 is complexed. Thus, it is more likely α-synuclein participates in the switching of Munc-13/18. To complete neurotransmitter release, the SNARE motifs of VAMP2, syntaxin-1, and SNAP25 (trans-SNARE-complex) yield a half-zipper conformation that is structurally aided by the synaptotagmin protein family and complexins (SNAREpins).
Each component of the SNARE complexes is individually regulated (i.e., syntaxin-1 with Munc-13-Munc-18, SNAP25 with CSPα-Hsc70, synaptotagmin with complexins, and VAMP2 with α-synuclein). Multiple layers of regulation are present for each component, with both synaptotagmin and α-synuclein being functionally calcium-dependent [64][65][66]. The requirement for α-synuclein to embed into lipid vesicles is not a primary target of regulation as it is the disequilibrium between lipid-bound and monomeric species that may propagate disease pathogenesis. In addition to the above function, α-synuclein modulates synaptic plasticity through enhancing dopamine reuptake from the presynaptic cleft. α-Synuclein-knockout mice exhibit minimal dysfunction in terms of neurotransmission but do have altered dopamine reuptake in the nigrostriatal pathway [67]. However, when silencing all three synuclein isoforms (α, β, and γ), mice exhibit significantly impaired membrane fusion and SNARE complex formation [38]. Silencing all synuclein isoforms spawned age-dependent dysfunction in vivo and in vitro, evidently mediating synaptic junctions to diminish by ~30% in size, retinal degeneration, increased VAMP2 expression, and poor survivability [68].
After exocytosis, α-synuclein modulates synaptic vesicle recycling and subsequently docks at dopamine transporters (DAT) via weak C-terminal interactions [69][70]. It remains unclear exactly how dopamine reuptake occurs, but docked vesicles bind DAT through the transmembrane protein synaptogyrin-3, facilitating dopamine–proton exchange [71]. As a result, dopamine reuptake is trafficked directly into synaptic vesicles, avoiding oxidative cytotoxicity. In addition to its synaptic role, α-synuclein may also participate in endoplasmic reticulum (ER) to trans-Golgi network (TGN) trafficking by interacting with RAS-related protein in brain 1a (Rab1a) [72]. The latter GTPase has been linked to macroautophagy and autophagosome trafficking, as shown by its overexpression rescuing cells from α-synuclein-dependent dysfunction of ferritinophagy in transfected retinal pigmented epithelial cells [73].
4. Iron and Calcium Regulation of α-Synuclein
Regulation of α-synuclein is almost entirely at the post-transcriptional level, depending upon numerous metal ions for functional activity. Up to eight calcium ions bind along the C-terminus at residues E104, A107, I112, D119, E123, A124, and E126 [37]. However, physiological scenarios only require six calcium atoms for functionality. In vitro, α-synuclein monomers can only interact with hydrophobic surfaces at its N-terminus, yet, when calcium was added to the suspension, hydrophobic interactions with phospholipid membranes were increased by 5-fold [48]. Calcium enhanced the hydrophobicity across the C-terminus and central non-amyloid component (NAC) domain, promoting membrane interaction [74].
4.1. Aggregation Induction of α-Synuclein
In vivo, voltage-gated calcium channels (VGCCs) can increase calcium concentrations to 200–300 µM in microdomains, exceeding the Kd of 21 µM for α-synuclein [37][75]. Furthermore, copper ions, bound at D2 and H50, support the folding of α-synuclein N-terminal helices [76]. Unlike other metals, the interactions between iron and α-synuclein are far less understood. On the one hand, reports have described α-synuclein as a ferrireductase, catalyzing the reduction of Fe3+ to Fe2+ [77]. While on the other hand, nuclear magnetic resonance (NMR) spectroscopy suggests that iron has a poor micromolar affinity and may only interact with the Asp121 residue [78][79].
Fibrillogenesis (i.e., the generation of amyloid-like fibril structures) stems from calcium exposure to monomeric α-synuclein, ultimately prompting hydrophobic interactions between monomeric species through β-sheet stacking [37][80]. Notably, the enhanced hydrophobicity amongst the NAC domain is the driving force behind fibril development [61][81]. However, the complete misfolding of monomeric α-synuclein can yield annular species that may lodge in lipid membranes and distort cellular permeability to ions [82]. In the context of toxicity, preformed fibrils can substantially impede cell viability at nanomolar concentrations, despite no endogenous expression of α-synuclein in certain cell types [10][83][84]. It has been suggested that the SNCA gene contains an atypical iron response element (IRE) in its 3′-untranslated region (3′-UTR) [85]. Such a theory could indicate that intracellular iron concentrations may promote α-synuclein expression. However, it is essential to note that this IRE may be non-functional, as it does not conform to typical canonical sequences that bind iron-response proteins (IRPs) [86]. Further studies are required to determine if this IRE plays any role in the regulation of α-synuclein expression by iron.
4.2. Iron-Induced Catecholamine Oxidation and Redox Damage
Iron metabolism is important when considering the marked redox potential of catecholamines. For example, iron can ligate to the adjacent hydroxyl groups of dopamine [6][87] (Figure 2). The resultant dopamine quinone can be further transformed into 6-hydroxy dopamine (6-OHDA) in the presence of hydrogen peroxide (H2O2) [88]. 6-OHDA is a potent neurotoxin used to replicate PD pathologies in mice, and unlike 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-OHDA specifically damages dopaminergic neurons through DAT-mediated uptake [88]. Aminochrome synthesis may occur directly from iron–dopamine adducts or as a downstream 6-OHDA product. At physiological pH, aminochrome is cytotoxic through redox cycling. The subsequent production of 6-dihydroxindol (neuromelanin) is neuroprotective, presumably via the chelation of redox-active iron [89]. Ultimately, neuromelanin seemingly suppresses redox cycling in acidic vesicles (e.g., synaptic vesicles, autophagosomes, and lysosomes) [88].
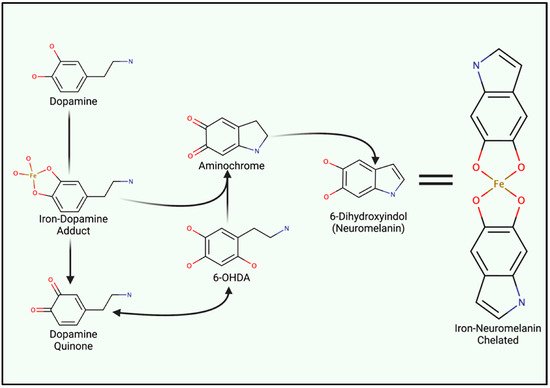
Figure 2. Iron Involvement in Dopamine Oxidation and Neuromelanin synthesis. The hydroxyl groups of dopamine can directly chelate iron, catalyzing the generation of either aminochrome or dopamine quinone. Both analogues induce mitochondrial dysfunction and oxidative stress. However, quinone species can undergo reactions to generate 6-hydroxydopamine (6-OHDA). Only differing by an additional hydroxyl group, 6-OHDA can enter cells through DAT to induce internal redox stress. Acidic vesicles provide a reducing environment for aminochrome carbonyl groups, ultimately yielding 6-dihydroindol (neuromelanin) which can form adducts between reactive ions.
Neuromelanin chelates iron and other reactive products via two hydroxyl groups upon the fifth and sixth carbon (Figure 2), yielding a benzothiazepine-like complex [8][88][90]. Redox-active iron and copper can directly oxidize dopamine; thus, both can induce neuromelanin synthesis and chelation. However, iron is by far the most prominent substrate [91]. Isolated neuromelanin contains ~11 μg/mg of iron, while basal tissue concentrations remain approximately 0.1–0.25 μg/mg [89][92]. Neuromelanin localizes to autophagosomes due to its autophagy-dependent clearance (melanophagy) [93]. Neuromelanin concentrations increase with age and in response to prolonged exposure to reactive species and lysosomal failure, giving rise to age-dependent pigmentation. Transmission electron microscopy of healthy substantia nigra and locus coeruleus tissue illustrates neuromelanin-positive autophagosomes that exceed 1 µm in diameter [93]. Vesicular clustering, increased metal concentrations, and autophagy-lysosomal dysfunction are both indicative of senescent cells as well as many neurodegenerative diseases (e.g., Alzheimer’s disease (AD), HD, and amyotrophic lateral sclerosis (ALS) and PD) [94][95][96][97][98].
5. Iron Entry, Regulation and Cellular Metabolism
Iron acquisition by cells can be separated into transferrin-dependent or transferrin-receptor-independent mechanisms. The transferrin receptor is not expressed in glia, yet these cells become burdened with high intracellular iron levels and ferritin in disease states [99][100]. Moreover, the CNS contains a high concentration of low molecular weight iron with some proportion being chelated and reduced to Fe2+ by ascorbate [101][102]. The mechanism of how iron loading occurs in dopaminergic neurons remains elusive. In recent years, VGCCs, particularly those of the L-type family, has been recognized as being permeable to iron (Fe2+), thus providing a potential path for unregulated iron entry and intrinsic redox damage [103].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23042378
References
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236.
- Watanabe, I.; Vachal, E.; Tomita, T. Dense core vesicles around the Lewy body in incidental Parkinson’s disease: An electron microscopic study. Acta Neuropathol. 1977, 39, 173–175.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm 2020, 127, 181–187.
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264.
- Abeyawardhane, D.L.; Lucas, H.R. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxid. Med. Cell Longev. 2019, 2019, 4609702.
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975.
- Zecca, L.; Shima, T.; Stroppolo, A.; Goj, C.; Battiston, G.A.; Gerbasi, R.; Sarna, T.; Swartz, H.M. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience 1996, 73, 407–415.
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542.
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7.
- Calo, L.; Wegrzynowicz, M.; Santivanez-Perez, J.; Grazia Spillantini, M. Synaptic failure and alpha-synuclein. Mov. Disord. 2016, 31, 169–177.
- Di Marco Vieira, B.; Radford, R.A.W.; Hayashi, J.; Eaton, E.D.; Greenaway, B.; Jambas, M.; Petcu, E.B.; Chung, R.S.; Pountney, D.L. Extracellular Alpha-Synuclein Promotes a Neuroinhibitory Secretory Phenotype in Astrocytes. Life 2020, 10, 183.
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.M.; Sobott, F. Metal ions shape alpha-synuclein. Sci. Rep. 2020, 10, 16293.
- Han, J.; Day, J.R.; Connor, J.R.; Beard, J.L. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J. Nutr. 2002, 132, 2769–2774.
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 2009, 24, 673–684.
- Meyron-Holtz, E.G.; Cohen, L.A.; Fahoum, L.; Haimovich, Y.; Lifshitz, L.; Magid-Gold, I.; Stuemler, T.; Truman-Rosentsvit, M. Ferritin polarization and iron transport across monolayer epithelial barriers in mammals. Front. Pharmacol. 2014, 5, 194.
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352.
- Everett, J.; Brooks, J.; Lermyte, F.; O’Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. Iron stored in ferritin is chemically reduced in the presence of aggregating Abeta(1-42). Sci. Rep. 2020, 10, 10332.
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114.
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018, 233, 9179–9190.
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238.
- Hodge, G.K.; Butcher, L.L. Pars compacta of the substantia nigra modulates motor activity but is not involved importantly in regulating food and water intake. Naunyn. Schmiedebergs Arch. Pharmacol. 1980, 313, 51–67.
- Fabbri, M.; Reimao, S.; Carvalho, M.; Nunes, R.G.; Abreu, D.; Guedes, L.C.; Bouca, R.; Lobo, P.P.; Godinho, C.; Coelho, M.; et al. Substantia Nigra Neuromelanin as an Imaging Biomarker of Disease Progression in Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 491–501.
- Liang, C.L.; Sinton, C.M.; Sonsalla, P.K.; German, D.C. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 1996, 5, 313–318.
- de Berker, A.O.; Rutledge, R.B. A role for the human substantia nigra in reinforcement learning. J. Neurosci. 2014, 34, 12947–12949.
- Galtieri, D.J.; Estep, C.M.; Wokosin, D.L.; Traynelis, S.; Surmeier, D.J. Pedunculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife 2017, 6, e30352.
- Sonne, J.; Reddy, V.; Beato, M.R. Neuroanatomy, Substantia Nigra. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152.
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829.
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926.
- Melland, H.; Carr, E.M.; Gordon, S.L. Disorders of synaptic vesicle fusion machinery. J. Neurochem. 2021, 157, 130–164.
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; O’Donnell, P.; Drobatz, K.; Gurda, B.; et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J. Pharmacol. Exp. Ther. 2016, 358, 254–261.
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thevenod, F. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145.
- Du, X.; Xu, H.; Shi, L.; Jiang, Z.; Song, N.; Jiang, H.; Xie, J. Activation of ATP-sensitive potassium channels enhances DMT1-mediated iron uptake in SK-N-SH cells in vitro. Sci. Rep. 2016, 6, 33674.
- Bazelon, M.; Fenichel, G.M.; Randall, J. Studies on neuromelanin. I. A melanin system in the human adult brainstem. Neurology 1967, 17, 512–519.
- Carballo-Carbajal, I.; Laguna, A.; Romero-Gimenez, J.; Cuadros, T.; Bove, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Penuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973.
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712.
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Trexler, A.J.; Rhoades, E. N-Terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 2012, 21, 601–605.
- Deng, S.; Pan, B.; Gottlieb, L.; Petersson, E.J.; Marmorstein, R. Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. Elife 2020, 9, e57491.
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124.
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449.
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475.
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173.
- Hong, W.; Lev, S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35–43.
- Hawk, B.J.D.; Khounlo, R.; Shin, Y.K. Alpha-Synuclein Continues to Enhance SNARE-Dependent Vesicle Docking at Exorbitant Concentrations. Front. Neurosci. 2019, 13, 216.
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115.
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Sudhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122.
- Koo, S.J.; Markovic, S.; Puchkov, D.; Mahrenholz, C.C.; Beceren-Braun, F.; Maritzen, T.; Dernedde, J.; Volkmer, R.; Oschkinat, H.; Haucke, V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. USA 2011, 108, 13540–13545.
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283.
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159.
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39.
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396.
- Sharma, M.; Burre, J.; Bronk, P.; Zhang, Y.; Xu, W.; Sudhof, T.C. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012, 31, 829–841.
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763.
- Zhou, P.; Pang, Z.P.; Yang, X.; Zhang, Y.; Rosenmund, C.; Bacaj, T.; Sudhof, T.C. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. 2013, 32, 159–171.
- Wang, S.; Li, Y.; Gong, J.; Ye, S.; Yang, X.; Zhang, R.; Ma, C. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat. Commun. 2019, 10, 69.
- Shao, K.; Li, F.; Yang, Y.; Wang, N.; Gao, X.D.; Nakanishi, H. Characteristics of SNARE proteins are defined by distinctive properties of SNARE motifs. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129658.
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Gotz, J.; Parton, R.G.; Collins, B.M.; et al. Munc18-1 is a molecular chaperone for alpha-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 2016, 214, 705–718.
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479.
- Lee, H.K.; Yang, Y.; Su, Z.; Hyeon, C.; Lee, T.S.; Lee, H.W.; Kweon, D.H.; Shin, Y.K.; Yoon, T.Y. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 2010, 328, 760–763.
- Zhou, Q.; Zhou, P.; Wang, A.L.; Wu, D.; Zhao, M.; Sudhof, T.C.; Brunger, A.T. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548, 420–425.
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252.
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578.
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554.
- Ishibashi, K.; Oda, K.; Ishiwata, K.; Ishii, K. Comparison of dopamine transporter decline in a patient with Parkinson’s disease and normal aging effect. J. Neurol. Sci. 2014, 339, 207–209.
- Egana, L.A.; Cuevas, R.A.; Baust, T.B.; Parra, L.A.; Leak, R.K.; Hochendoner, S.; Pena, K.; Quiroz, M.; Hong, W.C.; Dorostkar, M.M.; et al. Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-.-3. J. Neurosci. 2009, 29, 4592–4604.
- Cooper, A.A. Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328.
- Baksi, S.; Singh, N. alpha-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 2017, 7, 12843.
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and alpha-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 237.
- Llinas, R.; Sugimori, M.; Silver, R.B. Microdomains of high calcium concentration in a presynaptic terminal. Science 1992, 256, 677–679.
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, copper and alpha-Synuclein in alpha-synucleinopathies. Front. Neurosci. 2017, 11, 114.
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814.
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901.
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and alpha-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260.
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca(2+) Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004.
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902.
- Pountney, D.L.; Voelcker, N.H.; Gai, W.P. Annular alpha-synuclein oligomers are potentially toxic agents in alpha-synucleinopathy. Hypothesis. Neurotox. Res. 2005, 7, 59–67.
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar alpha-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513.
- Hindeya Gebreyesus, H.; Gebrehiwot Gebremichael, T. The Potential Role of Astrocytes in Parkinson’s Disease (PD). Med. Sci. 2020, 8, 7.
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5’-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 2007, 12, 222–223.
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896.
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035.
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119.
- Shima, T.; Sarna, T.; Swartz, H.M.; Stroppolo, A.; Gerbasi, R.; Zecca, L. Binding of iron to neuromelanin of human substantia nigra and synthetic melanin: An electron paramagnetic resonance spectroscopy study. Free Radic. Biol. Med. 1997, 23, 110–119.
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell Neurosci. 2020, 14, 218.
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848.
- Sulzer, D.; Cassidy, C.; Horga, G.; Kang, U.J.; Fahn, S.; Casella, L.; Pezzoli, G.; Langley, J.; Hu, X.P.; Zucca, F.A.; et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 11.
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030.
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614.
- del Toro, D.; Alberch, J.; Lazaro-Dieguez, F.; Martin-Ibanez, R.; Xifro, X.; Egea, G.; Canals, J.M. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 2009, 20, 1478–1492.
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590.
- Cho, S.J.; Yun, S.M.; Jo, C.; Lee, D.H.; Choi, K.J.; Song, J.C.; Park, S.I.; Kim, Y.J.; Koh, Y.H. SUMO1 promotes Abeta production via the modulation of autophagy. Autophagy 2015, 11, 100–112.
- Tadokoro, K.; Yamashita, T.; Shang, J.; Ohta, Y.; Nomura, E.; Morihara, R.; Omote, Y.; Takemoto, M.; Abe, K. Switching the Proteolytic System from the Ubiquitin-Proteasome System to Autophagy in the Spinal Cord of an Amyotrophic Lateral Sclerosis Mouse Model. Neuroscience 2021, 466, 47–57.
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281.
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339.
- Wilson, R.L.; Wightman, R.M. Systemic and Nigral Application of Amphetamine Both Cause an Increase in Extracellular Concentration of Ascorbate in the Caudate-Nucleus of the Rat. Brain Res. 1985, 339, 219–226.
- Schenk, J.O.; Miller, E.; Gaddis, R.; Adams, R.N. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982, 253, 353–356.
- Boag, M.K.; Ma, L.; Mellick, G.D.; Pountney, D.L.; Feng, Y.; Quinn, R.J.; Liew, A.W.-C.; Dharmasivam, M.; Azad, M.G.; Afroz, R.; et al. Calcium channels and iron metabolism: A redox catastrophe in Parkinson’s disease and an innovative path to novel therapies? Redox Biol. 2021, 47, 102136.
This entry is offline, you can click here to edit this entry!