 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gang-Jee Ko | + 1753 word(s) | 1753 | 2022-01-18 08:46:46 | | | |
| 2 | Jason Zhu | + 92 word(s) | 1845 | 2022-03-03 03:18:17 | | |
Video Upload Options
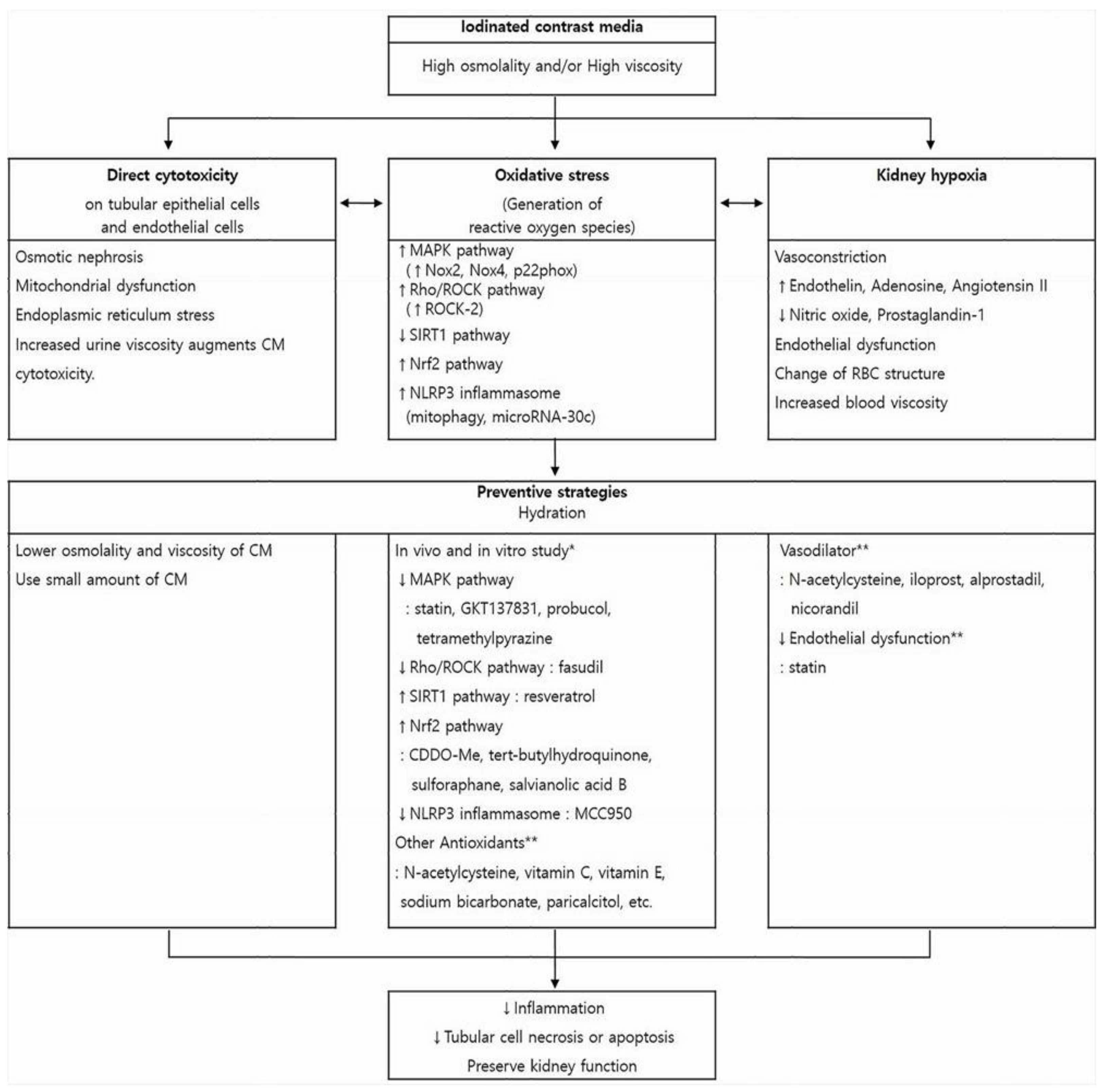
Contrast-induced nephropathy (CIN) is an impairment of renal function that occurs after the administration of an iodinated contrast medium (CM). Kidney dysfunction in CIN is considered transient and reversible in most cases. However, it is the third most common cause of hospital-acquired acute kidney injury and is associated with increased morbidity and mortality, especially in high-risk patients. Diagnostic and interventional procedures that require intravascular CM are being used with increasing frequency, especially among the elderly, who can be particularly susceptible to CIN due to multiple comorbidities. Therefore, identifying the exact mechanisms of CIN and its associated risk factors is crucial not only to provide optimal preventive management for at-risk patients, but also to increase the feasibility of diagnostic and interventional procedure that use CM.
1. Introduction
2. Pathophysiology
References
- Hou, S.H.; Bushinsky, D.A.; Wish, J.B.; Cohen, J.J.; Harrington, J.T. Hospital-acquired renal insufficiency: A prospective study. Am. J. Med. 1983, 74, 243–248.
- Nash, K.; Hafeez, A.; Hou, S. Hospital-acquired renal insufficiency. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2002, 39, 930–936.
- Abe, M.; Morimoto, T.; Akao, M.; Furukawa, Y.; Nakagawa, Y.; Shizuta, S.; Ehara, N.; Taniguchi, R.; Doi, T.; Nishiyama, K.; et al. Relation of contrast-induced nephropathy to long-term mortality after percutaneous coronary intervention. Am. J. Cardiol. 2014, 114, 362–368.
- Azzalini, L.; Kalra, S. Contrast-Induced Acute Kidney Injury-Definitions, Epidemiology, and Implications. Interv. Cardiol. Clin. 2020, 9, 299–309.
- Haveman, J.W.; Gansevoort, R.T.; Bongaerts, A.H.; Nijsten, M.W. Low incidence of nephropathy in surgical ICU patients receiving intravenous contrast: A retrospective analysis. Intensive Care Med. 2006, 32, 1199–1205.
- Kooiman, J.; Pasha, S.M.; Zondag, W.; Sijpkens, Y.W.; van der Molen, A.J.; Huisman, M.V.; Dekkers, O.M. Meta-analysis: Serum creatinine changes following contrast enhanced CT imaging. Eur. J. Radiol. 2012, 81, 2554–2561.
- Chousterman, B.G.; Bouadma, L.; Moutereau, S.; Loric, S.; Alvarez-Gonzalez, A.; Mekontso-Dessap, A.; Laissy, J.P.; Rahmouni, A.; Katsahian, S.; Brochard, L.; et al. Prevention of contrast-induced nephropathy by N-acetylcysteine in critically ill patients: Different definitions, different results. J. Crit. Care 2013, 28, 701–709.
- Mamoulakis, C.; Tsarouhas, K.; Fragkiadoulaki, I.; Heretis, I.; Wilks, M.F.; Spandidos, D.A.; Tsitsimpikou, C.; Tsatsakis, A. Contrast-induced nephropathy: Basic concepts, pathophysiological implications and prevention strategies. Pharmacol. Ther. 2017, 180, 99–112.
- Faucon, A.L.; Bobrie, G.; Clement, O. Nephrotoxicity of iodinated contrast media: From pathophysiology to prevention strategies. Eur. J. Radiol. 2019, 116, 231–241.
- Ward, D.B.; Valentovic, M.A. Contrast Induced Acute Kidney Injury and Direct Cytotoxicity of Iodinated Radiocontrast Media on Renal Proximal Tubule Cells. J. Pharmacol. Exp. Ther. 2019, 370, 160–171.
- Mehran, R.; Dangas, G.D.; Weisbord, S.D. Contrast-Associated Acute Kidney Injury. N. Engl. J. Med. 2019, 380, 2146–2155.
- McCullough, P.A.; Choi, J.P.; Feghali, G.A.; Schussler, J.M.; Stoler, R.M.; Vallabahn, R.C.; Mehta, A. Contrast-Induced Acute Kidney Injury. J. Am. Coll. Cardiol. 2016, 68, 1465–1473.
- Caiazza, A.; Russo, L.; Sabbatini, M.; Russo, D. Hemodynamic and tubular changes induced by contrast media. BioMed Res. Int. 2014, 2014, 578974.
- Liu, Z.Z.; Viegas, V.U.; Perlewitz, A.; Lai, E.Y.; Persson, P.B.; Patzak, A.; Sendeski, M.M. Iodinated contrast media differentially affect afferent and efferent arteriolar tone and reactivity in mice: A possible explanation for reduced glomerular filtration rate. Radiology 2012, 265, 762–771.
- Heyman, S.N.; Khamaisi, M.; Zorbavel, D.; Rosen, S.; Abassi, Z. Role of Hypoxia in Renal Failure Caused by Nephrotoxins and Hypertonic Solutions. Semin. Nephrol. 2019, 39, 530–542.
- Leisman, S. Radiocontrast Toxicity. Adv. Chronic Kidney Dis. 2020, 27, 50–55.
- Grainger, R.G. Osmolality of intravascular radiological contrast media. Br. J. Radiol. 1980, 53, 739–746.
- Sendeski, M.M.; Persson, A.B.; Liu, Z.Z.; Busch, J.F.; Weikert, S.; Persson, P.B.; Hippenstiel, S.; Patzak, A. Iodinated contrast media cause endothelial damage leading to vasoconstriction of human and rat vasa recta. Am. J. Physiol. Ren. Physiol. 2012, 303, F1592–F1598.
- Liu, Z.Z.; Schmerbach, K.; Lu, Y.; Perlewitz, A.; Nikitina, T.; Cantow, K.; Seeliger, E.; Persson, P.B.; Patzak, A.; Liu, R.; et al. Iodinated contrast media cause direct tubular cell damage, leading to oxidative stress, low nitric oxide, and impairment of tubuloglomerular feedback. Am. J. Physiol. Ren. Physiol. 2014, 306, F864–F872.
- Lin, H.H.; Lee, T.S.; Lin, S.J.; Yeh, Y.C.; Lu, T.M.; Hsu, C.P. DDAH-2 alleviates contrast medium iopromide-induced acute kidney injury through nitric oxide synthase. Clin. Sci. 2019, 133, 2361–2378.
- Kusirisin, P.; Chattipakorn, S.C.; Chattipakorn, N. Contrast-induced nephropathy and oxidative stress: Mechanistic insights for better interventional approaches. J. Transl. Med. 2020, 18, 400.
- Pisani, A.; Riccio, E.; Andreucci, M.; Faga, T.; Ashour, M.; Di Nuzzi, A.; Mancini, A.; Sabbatini, M. Role of reactive oxygen species in pathogenesis of radiocontrast-induced nephropathy. BioMed Res. Int. 2013, 2013, 868321.
- Jeong, B.Y.; Lee, H.Y.; Park, C.G.; Kang, J.; Yu, S.L.; Choi, D.R.; Han, S.Y.; Park, M.H.; Cho, S.; Lee, S.Y.; et al. Oxidative stress caused by activation of NADPH oxidase 4 promotes contrast-induced acute kidney injury. PLoS ONE 2018, 13, e0191034.
- Liu, G.L.; Lei, R.; Duan, S.B.; Tang, M.M.; Luo, M.; Xu, Q. Atorvastatin alleviates iodinated contrast media-induced cytotoxicity in human proximal renal tubular epithelial cells. Exp. Ther. Med. 2017, 14, 3309–3313.
- Gong, X.; Wang, Q.; Tang, X.; Wang, Y.; Fu, D.; Lu, H.; Wang, G.; Norgren, S. Tetramethylpyrazine prevents contrast-induced nephropathy by inhibiting p38 MAPK and FoxO1 signaling pathways. Am. J. Nephrol. 2013, 37, 199–207.
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554.
- Jin, L.; Ying, Z.; Webb, R.C. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1495–H1500.
- Wang, Y.; Zhang, H.; Yang, Z.; Miao, D.; Zhang, D. Rho Kinase Inhibitor, Fasudil, Attenuates Contrast-induced Acute Kidney Injury. Basic Clin. Pharmacol. Toxicol. 2018, 122, 278–287.
- Khaleel, S.A.; Raslan, N.A.; Alzokaky, A.A.; Ewees, M.G.; Ashour, A.A.; Abdel-Hamied, H.E.; Abd-Allah, A.R. Contrast media (meglumine diatrizoate) aggravates renal inflammation, oxidative DNA damage and apoptosis in diabetic rats which is restored by sulforaphane through Nrf2/HO-1 reactivation. Chem.-Biol. Interact. 2019, 309, 108689.
- Hong, Y.A.; Bae, S.Y.; Ahn, S.Y.; Kim, J.; Kwon, Y.J.; Jung, W.Y.; Ko, G.J. Resveratrol Ameliorates Contrast Induced Nephropathy Through the Activation of SIRT1-PGC-1alpha-Foxo1 Signaling in Mice. Kidney Blood Press. Res. 2017, 42, 641–653.
- Kim, J.E.; Bae, S.Y.; Ahn, S.Y.; Kwon, Y.J.; Ko, G.J. The role of nuclear factor erythroid-2-related factor 2 expression in radiocontrast-induced nephropathy. Sci. Rep. 2019, 9, 2608.
- Zhao, Z.; Liao, G.; Zhou, Q.; Lv, D.; Holthfer, H.; Zou, H. Sulforaphane Attenuates Contrast-Induced Nephropathy in Rats via Nrf2/HO-1 Pathway. Oxidative Med. Cell. Longev. 2016, 2016, 9825623.
- Zhou, Q.; Wang, X.; Shao, X.; Wang, H.; Liu, X.; Ke, X.; Xiong, C.; Wei, L.; Zou, H. tert-Butylhydroquinone Treatment Alleviates Contrast-Induced Nephropathy in Rats by Activating the Nrf2/Sirt3/SOD2 Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 4657651.
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. JASN 2018, 29, 1799–1809.
- Gao, D.; Wang, H.; Xu, Y.; Zheng, D.; Zhang, Q.; Li, W. Protective effect of astaxanthin against contrast-induced acute kidney injury via SIRT1-p53 pathway in rats. Int. Urol. Nephrol. 2019, 51, 351–358.
- Wang, Y.; Wang, B.; Qi, X.; Zhang, X.; Ren, K. Resveratrol Protects Against Post-Contrast Acute Kidney Injury in Rabbits With Diabetic Nephropathy. Front. Pharmacol. 2019, 10, 833.
- Tongqiang, L.; Shaopeng, L.; Xiaofang, Y.; Nana, S.; Xialian, X.; Jiachang, H.; Ting, Z.; Xiaoqiang, D. Salvianolic Acid B Prevents Iodinated Contrast Media-Induced Acute Renal Injury in Rats via the PI3K/Akt/Nrf2 Pathway. Oxidative Med. Cell. Longev. 2016, 2016, 7079487.
- Song, L.; Yao, S.; Zheng, D.; Xuan, Y.; Li, W. Astaxanthin attenuates contrast-induced acute kidney injury in rats via ROS/NLRP3 inflammasome. Int. Urol. Nephrol. 2021.
- Cheng, W.; Zhao, F.; Tang, C.Y.; Li, X.W.; Luo, M.; Duan, S.B. Comparison of iohexol and iodixanol induced nephrotoxicity, mitochondrial damage and mitophagy in a new contrast-induced acute kidney injury rat model. Arch. Toxicol. 2018, 92, 2245–2257.
- Shen, J.; Wang, L.; Jiang, N.; Mou, S.; Zhang, M.; Gu, L.; Shao, X.; Wang, Q.; Qi, C.; Li, S.; et al. NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci. Rep. 2016, 6, 34682.
- Lin, Q.; Li, S.; Jiang, N.; Jin, H.; Shao, X.; Zhu, X.; Wu, J.; Zhang, M.; Zhang, Z.; Shen, J.; et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 2021, 17, 2975–2990.
- Tan, X.; Zheng, X.; Huang, Z.; Lin, J.; Xie, C.; Lin, Y. Involvement of S100A8/A9-TLR4-NLRP3 Inflammasome Pathway in Contrast-Induced Acute Kidney Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 209–222.
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254.
- Xu, J.; Ma, L.; Fu, P. MicroRNA-30c attenuates contrast-induced acute kidney injury by suppressing NLRP3 inflammasome. Int. Immunopharmacol. 2020, 87, 106457.

