Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Urology & Nephrology
Contrast-induced nephropathy (CIN) is an impairment of renal function that occurs after the administration of an iodinated contrast medium (CM). Kidney dysfunction in CIN is considered transient and reversible in most cases.
- Pathophysiology
- Contrast-induced nephropathy
- contrast medium (CM)
1. Introduction
Contrast-induced nephropathy (CIN) is an impairment of kidney function that occurs after the administration of iodinated contrast medium (CM). It is the third most common cause of hospital-acquired acute kidney injury (AKI) and is associated with prolonged hospital stay and increased morbidity and mortality [1,2,3]. The reported incidence of CIN varies from <1% to greater than 50% depending on patient risk factors, type of procedure, and definition of CIN [4,5,6,7]. Diagnostic and interventional procedures that require intravascular CM are being used with increasing frequency, especially among the elderly, who can be particularly susceptible to CIN due to multiple comorbidities such as chronic kidney disease (CKD) and diabetes mellitus. Therefore, it is important to understand the precise risks and pathophysiology of CIN to provide optimal preventive management.
2. Pathophysiology
The exact pathophysiology of CIN is not fully understood. Direct cytotoxicity, altered intrarenal hemodynamics, and ROS generation have been proposed as the main pathophysiologic mechanisms of CIN [52]. Those three mechanisms influence and aggravate one another, creating a vicious cycle that ultimately leads to inflammation, tubular cell apoptosis, and impaired kidney function (Figure 1).
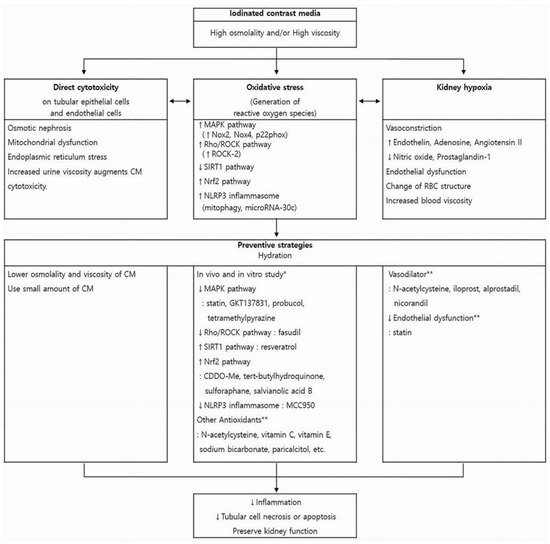
Figure 1. Pathophysiology of contrast-induced nephropathy (CIN) and promising strategies to preserve kidney function. Iodinated CM has direct cytotoxic effect on endothelial cells and renal tubular epithelial cells, induces vasoconstriction causing hypoxia in the outer medulla, and enhances the generation of reactive oxygen species. These changes influence one another and ultimately lead to kidney injury. Each box contains underlying mechanisms relevant to those three pathways. Hydration is the mainstay of CIN preventive strategies and can reduce harmful effect of CM in all three aspects. Other previously reported preventive measures and pharmaceutical agents are presented with regard to each pathogenic process. * These pharmaceutical agents have been studied in in vitro and in vivo experiments to reduce oxidative stress, that is, to reverse each pathogenic pathway. However, because Nrf2 expression increases during CM-induced oxidative stress as a cytoprotective response, Nrf2 activation is preventive against CIN. ** The preventive role of these agents on CIN is controversial. CM, contrast media; MAPK, mitogen-activated protein kinase; Nox, nicotinamide adenine dinucleotide phosphate oxidase; ROCK, rho-kinase; SIRT1, silent information regulator 1; Nrf2, nuclear factor erythroid 2-related factor 2; RBC, red blood cell.
CM has a direct cytotoxic effect on kidney tubular epithelial cells and vascular endothelial cells [35]. All types of contrast medium (CM) showed cytotoxic effects in vitro [52]. CM induced vacuolization in kidney tubular cells by pinocytosis (osmotic nephrosis), mitochondrial dysfunction that led to ROS generation and apoptosis, and endoplasmic reticulum stress that activated intrinsic apoptotic pathways [53]. Loss of the tubular brush border and cell membrane integrity and sloughing of the tubular epithelial cells into the lumen were caused by the direct cytotoxicity of CM [33,54].
CM administration induces transient vasodilation followed by vasoconstriction that can be sustained for several hours in the kidney vasculature as a result of alterations in kidney vasomodulators such as endothelin, adenosine, and NO [53,55]. Vasoconstriction of afferent arterioles reduces GFR and kidney blood flow, causing kidney parenchymal hypoxia [56]. The kidney outer medulla is in a relative hypoxic situation because of tubular ion transport in the basal state and the low partial pressure of oxygen with limited blood flow caused by the unique anatomy of the kidney vasculature. Hence, the thick ascending limbs of the loop of Henle (TAL) and segments of the proximal kidney tubules in the outer medulla are particularly susceptible to hypoxic injury [57].
The high osmolality and viscosity of CM causes osmotic diuresis, an increase in tubular pressure, and decrease in tubular and blood flow rates, all of which lead to an increase in tubular oxygen demand and a decrease in kidney blood supply [58]. Furthermore, CM induces direct vasoconstriction of the vasa recta through endothelial dysfunction and changes in red blood cell structure and function, both of which worsen kidney medullary hypoxia [34,59]. This mismatch between the metabolic demands of the TAL and the kidney medullary blood supply leads to oxidative stress. Tubular transport is associated with ROS production and the dense mitochondrial population in the proximal tubule and TAL is the major source of ROS [52]. Moreover, CM retention in the tubular lumen caused by the decreased tubular flow rate augments its cytotoxic impact. Ischemic and cytotoxic tubular cell damage again induces tubuloglomerular feedback, which enhances vasoconstriction of the afferent arteriole and produces further decreases in kidney blood flow and GFR [60].
The increase in ROS generation after exposure to CM has been observed in various in vitro and in vivo studies and can be explained partly by the diminished availability or activity of cellular antioxidant systems [61,62]. As explained above, both the direct cytotoxic effects of CM on tubular cells and kidney medullary hypoxia caused by vasoconstriction enhance ROS generation. Subsequently, ROS constrict kidney microcirculation and affect kidney vascular tone by modulating vasoactive substances such as NO [63]. In addition, oxidative DNA damage and multiple intracellular signaling pathways related to ROS lead to necrosis or apoptosis of kidney tubular cells. Because ROS is considered to play a central role in the pathogenesis of CIN, current research focuses on its involvement in CIN, either to elucidate the pathogenesis mechanism or to find an effective preventive or therapeutic target.
With regard to the pathophysiologic role of ROS in CIN, Kusirisin et al. reviewed in vitro and in vivo reports from PubMed up to September 2019 [62]. They summarized the intracellular signaling mechanisms associated with ROS in four pathways: (1) the mitogen-activated protein kinase (MAPK) pathway, which includes extracellular signal-related kinases, c-JUN N-terminal kinase, and p38; (2) the silent information regulator 1(SIRT1) pathway, which includes SIRT1, forkhead box type O transcription factors(FoxO), nuclear factor-κB(NF-κB), peroxisome proliferator-activated receptor gamma-assisted activating factor-1(PGC-1), and p53; (3) the Rho/Rho-kinase(Rho/ROCK) pathway, which includes myosin phosphatase target subunit 1 and NF-κB; (4) the nuclear factor erythroid2-related factor 2/heme oxygenase 1(Nrf-2/HO-1) pathway, which includes Nrf-2, nicotinamide adenine dinucleotide phosphate quinone oxidoreductase 1, glutathione, and HO-1.
CM increased ROS generation by upregulating nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2), Nox4, and p22phox, which led to apoptosis through the MAPK pathway [64,65,66]. ROCK belongs to the AGC (protein kinase A/protein kinase G/protein kinase C) family of serine/threonine kinases, which is a downstream target of the small GTPase Rho [67]. The Rho/ROCK pathway was reported to be activated by ROS [68]. ROCK-2 activity increases in CIN and regulates inflammation in the kidney [69]. Inhibiting the Rho/ROCK pathway decreased inflammation, the intracellular ROS level, and kidney cell apoptosis in mice with CIN and also induced kidney vasodilation and increased kidney artery blood flow [69].
Both the SIRT1-mediated and Nrf2-mediated pathways are involved in renoprotection against CM-induced oxidative stress and kidney cell apoptosis. SIRT1 decreased after exposure to CM, but Nrf2 expression increased during CM-induced oxidative stress as a cytoprotective response [70,71,72,73,74]. Activating either SIRT1 or Nrf2 attenuated CIN via diverse downstream mechanisms.
Sirtuins belong to a conserved family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases that is involved in multiple cellular functions related to proliferation, DNA repair, mitochondrial energy homeostasis, and antioxidant activity [75]. SIRT1 is the most widely studied sirtuin and is located in the nucleus, where it regulates both nucleosome histone acetylation and the activity of a variety of transcriptional factors and cofactors, including NF-κB, p53, FoxO, hypoxia-inducible factor(HIF)-2α, and PGC-1α [71,76]. Hong et al. examined the protective role of SIRT1 in CIN using NRK-52E cells and mice [71]. Iohexol decreased SIRT1 and PGC-1α expression both in vivo and in vitro. Using resveratrol to activate SIRT1 reduced oxidative stress, inflammation, and tubular cell apoptosis in mouse kidneys and increased the expression of SIRT1, PGC-1α, and dephosphorylated FoxO1 (activated form). Likewise, using siRNA to inhibit SIRT1 accentuated the decrease in NRK-52E cell viability after iohexol treatment. PGC-1α increased mitochondrial superoxide dismutase (SOD2) level and attenuated oxidative stress. Thus, the SIRT1-PGC-1α-Foxo1 signaling pathway was found to play a role in the development of CIN in mice. Wang et al. examined the involvement of the SIRT1–PGC-1α–HIF-1α signaling pathway in CIN using a rabbit model of diabetic nephropathy (DN rabbits) and HK-2 cells [77]. Resveratrol, a SIRT1 activator, inhibited iohexol-induced HK-2 cell apoptosis, which was enhanced by treatment with 2-MeOE2 (a HIF-1α inhibitor) under high-glucose conditions. In DN rabbits, SIRT1 activation was associated with the upregulation of PGC-1α and downregulation of HIF-1α, Bax, cleaved caspase-3, and cytochrome C protein. This was further verified in HK-2 cells under high-glucose conditions via 2-MeOE2 and SIRT1 inhibition using Ex527.
Nrf-2, a transcription factor, stimulates the transcription of genes that encode detoxifying and antioxidant enzymes, and increased Nrf-2 expression was noted as a cytoprotective response after exposure to CM [70,73,78]. Kim et al. evaluated the role of Nrf-2 in CIN using Nrf2 knockout mice and NRK-52E cells [72]. Loss of Nrf-2 function enhanced ROS production, inflammation, and apoptosis after iohexol treatment, whereas Nrf-2 activation via CDDO-Me co-treatment with iohexol attenuated tubular cell injury. Zhou et al. used a rat model of CIN and Nrf2-silenced HK-2 cells to reveal that the protective role of Nrf2 in CIN is mediated by the Nrf2/Sirt3/SOD2 signaling pathway [74]. SIRT3, a NAD+-dependent deacetylase localized in the mitochondrial matrix, regulates a variety of cellular processes and maintains mitochondrial function. SIRT3 protects against oxidative stress by transforming acetylated SOD2 into SOD2. Nrf2 activation using tert-butylhydroquinone reduced oxidative stress and kidney injury and increased SIRT3 and SOD2 expression in CIN rats. The Nrf2-mediated SIRT3/SOD2 pathway was validated in vitro. The expression of SIRT3 and SOD2 increased in HK-2 cells but decreased in Nrf2-silenced cells after ioversol treatment.
Apart from those four pathways, ROS-related mechanisms involving the NLRP3 inflammasome have been studied [79,80,81,82,83]. The NLRP3 inflammasome is associated with inflammation and apoptosis during AKI. Tan et al. demonstrated the involvement of the S100A8/A9-TLR4-NLRP3 inflammasome pathway in the development of CIN using rats with CIN and NRK-52E cells [83]. Lin et al. reported that PINK1-Parkin-mediated mitophagy protected kidney tubular epithelial cells by decreasing mitochondrial ROS and inhibiting the NLRP3 inflammasome [84]. Xu et al. revealed that the protective effect against CIN offered by microRNA-30c, which is upregulated under contrast exposure, is mediated by suppression of the NLRP3 inflammasome [85]. Attenuating CIN by directly inhibiting NLRP3 was demonstrated in in vivo (nlrp3 or casp1 knockout mice) and in vitro (treatment with MCC950, a selective NLRP3 inflammasome inhibitor) experiments that also resulted in the upregulation of HIF-1α and BNIP3-mediated mitophagy [82].
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics12010180
This entry is offline, you can click here to edit this entry!