 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jetty Chung-Yung Lee | + 3513 word(s) | 3513 | 2022-02-21 07:40:43 | | | |
| 2 | Jason Zhu | -9 word(s) | 3504 | 2022-03-02 03:31:01 | | | | |
| 3 | Jason Zhu | Meta information modification | 3504 | 2022-03-02 03:33:08 | | |
Video Upload Options
Vitamin E is one of the most popular fat-soluble vitamins in pathological research and has been under scrutiny since the 1980s as a vital dietary component of food. The antioxidant effect of vitamin E has been widely studied due to its benefits in the prevention of various cardiovascular diseases. The earliest research on vitamin E established its roles as a fat-soluble antioxidant due to lowered rates of atherosclerosis and an overall reduction in cardiovascular mortality observed in randomized-controlled trials. While these studies could not pinpoint the exact reasons behind vitamin E’s effect, it has been well-established in the past to be an effective radical scavenger to prevent LDL oxidation and foam cell formation, and subsequently prevents the formation of atherosclerotic lesions, inhibits plaque build-up and stenosis, and lowers hypertension—which are all major risk factors of poor vascular health.
1. Antioxidant Mechanism
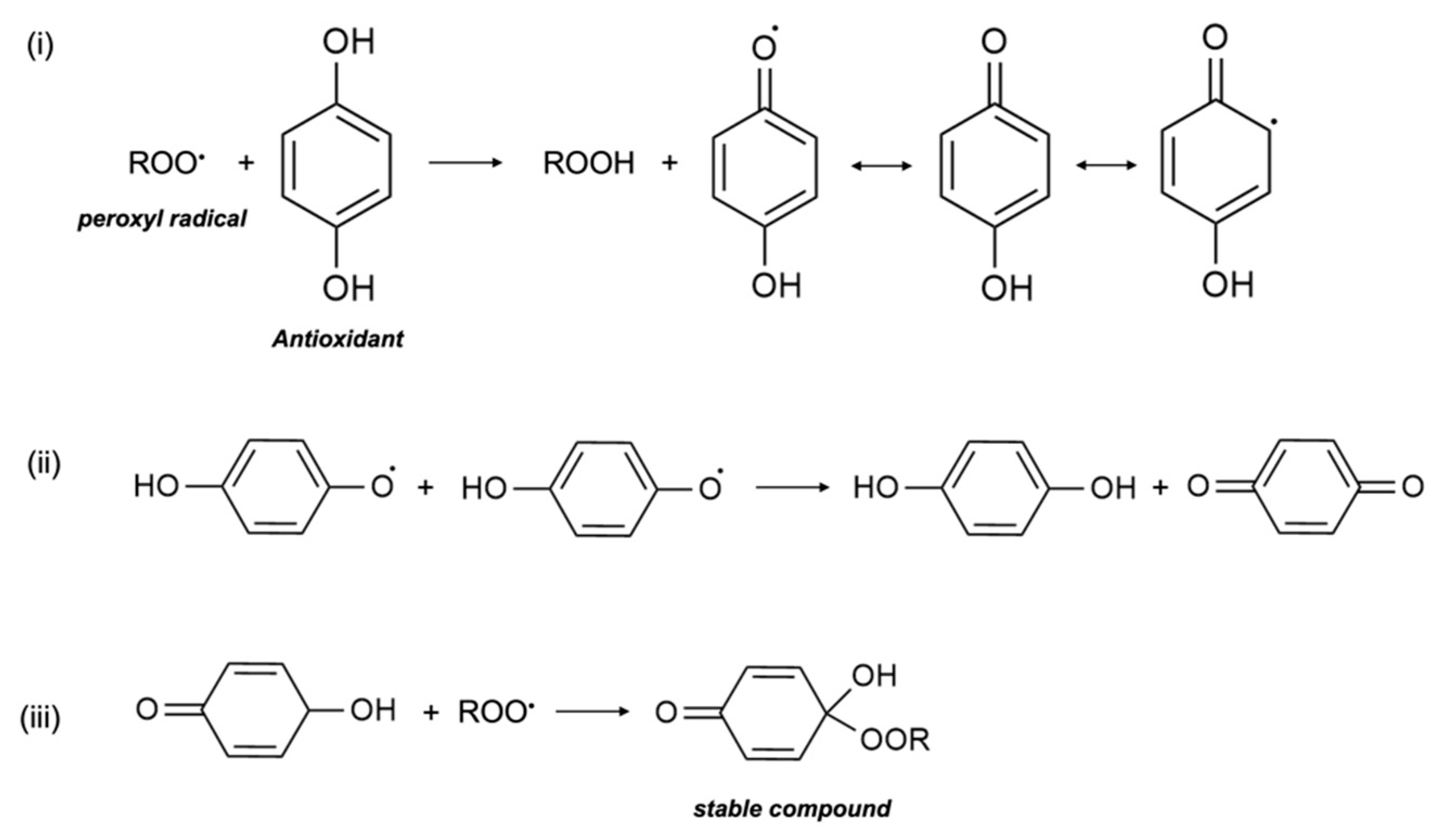
2. Potential Molecular Mechanisms
References
- Khadangi, F.; Azzi, A. Vitamin E—The Next 100 Years. IUBMB Life 2019, 71, 411–415.
- Traber, M.G.; Head, B. Vitamin E: How much is enough, too much and why! Free Radic. Biol. Med. 2021, 177, 212–225.
- Niki, E. Lipid oxidation that is, and is not, inhibited by vitamin E: Consideration about physiological functions of vitamin E. Free Radic. Biol. Med. 2021, 176, 1–15.
- Food, N.B.; Board, N. Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium and Carotenoids; National Academy Press: Washington, DC, USA, 2000.
- Diaz, M.N.; Frei, B.; Vita, J.A.; Keaney, J.F. Antioxidants and Atherosclerotic Heart Disease. N. Engl. J. Med. 1997, 337, 408–416.
- Witztum, J.L.; Steinberg, D. The Oxidative Modification Hypothesis of Atherosclerosis: Does It Hold for Humans? Trends Cardiovasc. Med. 2001, 11, 93–102.
- Stampfer, M.J.; Hennekens, C.H.; Manson, J.E.; Colditz, G.A.; Rosner, B.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Disease in Women. N. Engl. J. Med. 1993, 328, 1444–1449.
- Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Giovannucci, E.; Colditz, G.A.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Heart Disease in Men. N. Engl. J. Med. 1993, 328, 1450–1456.
- Hodis, H.N.; Mack, W.J.; LaBree, L.; Cashin-Hemphill, L.; Sevanian, A.; Johnson, R.; Azen, S.P. Serial Coronary Angiographic Evidence That Antioxidant Vitamin Intake Reduces Progression of Coronary Artery Atherosclerosis. JAMA 1995, 273, 1849–1854.
- Stephens, N.G.; Parsons, A.; Brown, M.J.; Schofield, P.M.; Kelly, F.; Cheeseman, K.; Mitchinson, M. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996, 347, 781–786.
- Keaney, J.F.; Guo, Y.; Cunningham, D.; Shwaery, G.T.; Xu, A.; Vita, J.A. Vascular incorporation of alpha-tocopherol prevents endothelial dysfunction due to oxidized LDL by inhibiting protein kinase C stimulation. J. Clin. Investig. 1996, 98, 386–394.
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218.
- Isakov, V.A.; Bogdanova, A.A.; Bessonov, V.V.; Sentsova, T.B.; Tutelyan, V.A.; Lin, Y.; Kazlova, V.; Hong, J.; Velliquette, R.A. Effects of Multivitamin, Multimineral and Phytonutrient Supplementation on Nutrient Status and Biomarkers of Heart Health Risk in a Russian Population: A Randomized, Double Blind, Placebo Controlled Study. Nutrients 2018, 10, 120.
- Alshiek, J.A.; Dayan, L.; Asleh, R.; Blum, S.; Levy, A.P.; Jacob, G. Anti-oxidative treatment with vitamin E improves peripheral vascular function in patients with diabetes mellitus and Haptoglobin 2-2 genotype: A double-blinded cross-over study. Diabetes Res. Clin. Pract. 2017, 131, 200–207.
- Bramley, P.M.; Elmadfa, I.; Kafatos, A.; Kelly, F.J.; Manios, Y.; Roxborough, H.E.; Schuch, W.; Sheehy, P.J.A.; Wagner, K.-H. Vitamin E. J. Sci. Food Agric. 2000, 80, 913–938.
- The HOPE and HOPE-TOO Trial Investigators. Effects of Long-term Vitamin E Supplementation on Cardiovascular Events and CancerA Randomized Controlled Trial. JAMA 2005, 293, 1338–1347.
- Lee, I.-M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the Primary Prevention of Cardiovascular Disease and CancerThe Women’s Health Study: A Randomized Controlled Trial. JAMA 2005, 294, 56–65.
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133.
- Collins, R. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes : a randomised placebo-controlled trial. Lancet 2002, 360, 23–33.
- Dalan, R.; Goh, L.L.; Lim, C.J.; Seneviratna, A.; Liew, H.; Seow, C.J.; Xia, L.; Chew, D.E.K.; Leow, M.K.S.; Boehm, B.O. Impact of Vitamin E supplementation on vascular function in haptoglobin genotype stratified diabetes patients (EVAS Trial): A randomised controlled trial. Nutr. Diabetes 2020, 10, 13.
- Brigelius-Flohé, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med. 2009, 46, 543–554.
- Howard, A.C.; McNeil, A.K.; McNeil, P.L. Promotion of plasma membrane repair by vitamin E. Nat. Commun. 2011, 2, 597.
- Chan, A.C.; Wagner, M.; Kennedy, C.; Chen, E.; Lanuville, O.; Mezl, V.A.; Tran, K.; Choy, P.C. Vitamin E up-regulates arachidonic acid releaseand phospholipase A2 in megakaryocytes. Mol. Cell. Biochem. 1998, 189, 153–159.
- Wong, M.; Lodge, J.K. A metabolomic investigation of the effects of vitamin E supplementation in humans. Nutr. Metab. 2012, 9, 110.
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336.
- Landes, N.; Pfluger, P.; Kluth, D.; Birringer, M.; Rühl, R.; Böl, G.-F.; Glatt, H.; Brigelius-Flohé, R. Vitamin E activates gene expression via the pregnane X receptor. Biochem. Pharmacol. 2003, 65, 269–273.
- Caputo, M.; Eletto, D.; Torino, G.; Tecce, M.F. Cooperation of docosahexaenoic acid and vitamin E in the regulation of UDP-glucuronosyltransferase mRNA expression. J. Cell. Physiol. 2008, 215, 765–770.
- Uemura, M.; Manabe, H.; Yoshida, N.; Fujita, N.; Ochiai, J.; Matsumoto, N.; Takagi, T.; Naito, Y.; Yoshikawa, T. α-Tocopherol prevents apoptosis of vascular endothelial cells via a mechanism exceeding that of mere antioxidation. Eur. J. Pharmacol. 2002, 456, 29–37.
- Nakamura, Y.K.; Omaye, S.T. α-Tocopherol modulates human umbilical vein endothelial cell expression of Cu/Zn superoxide dismutase and catalase and lipid peroxidation. Nutr. Res. 2008, 28, 671–680.
- Wallert, M.; Schmölz, L.; Galli, F.; Birringer, M.; Lorkowski, S. Regulatory metabolites of vitamin E and their putative relevance for atherogenesis. Redox Biol. 2014, 2, 495–503.
- Brigelius-Flohé, R. Vitamin E research: Past, now and future. Free Radic. Biol. Med. 2021, 177, 381–390.
- Tang, F.; Lu, M.; Zhang, S.; Mei, M.; Wang, T.; Liu, P.; Wang, H. Vitamin E Conditionally Inhibits Atherosclerosis in ApoE Knockout Mice by Anti-oxidation and Regulation of Vasculature Gene Expressions. Lipids 2014, 49, 1215–1223.
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J. Biol. Chem. 1991, 266, 6188–6194.
- Chatelain, E.; Boscoboinik, D.O.; Bartoli, G.-M.; Kagan, V.E.; Gey, F.K.; Packer, L.; Azzi, A. Inhibition of smooth muscle cell proliferation and protein kinase C activity by tocopherols and tocotrienols. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 1993, 1176, 83–89.
- Zingg, J.M. Vitamin E: Regulatory Role on Signal Transduction. IUBMB Life 2019, 71, 456–478.
- Tran, K.; Proulx, P.R.; Chan, A.C. Vitamin E suppresses diacylglycerol (DAG) level in thrombin-stimulated endothelial cells through an increase of DAG kinase activity. Biochim. Biophys. Acta (BBA)—Lipids Lipid Metab. 1994, 1212, 193–202.
- Hashemi, Z.; Sharifi, N.; Khani, B.; Aghadavod, E.; Asemi, Z. The effects of vitamin E supplementation on endometrial thickness, and gene expression of vascular endothelial growth factor and inflammatory cytokines among women with implantation failure. J. Matern.—Fetal Neonatal Med. 2019, 32, 95–102.
- Villacorta, L.; GraçA-Souza, A.V.; Ricciarelli, R.; Zingg, J.-M.; Azzi, A. α-Tocopherol Induces Expression of Connective Tissue Growth Factor and Antagonizes Tumor Necrosis Factor-α–Mediated Downregulation in Human Smooth Muscle Cells. Circ. Res. 2003, 92, 104–110.
- Chin, J.H.; Azhar, S.; Hoffman, B.B. Inactivation of endothelial derived relaxing factor by oxidized lipoproteins. J. Clin. Investig. 1992, 89, 10–18.
- Cachia, O.; Benna, J.E.; Pedruzzi, E.; Descomps, B.; Gougerot-Pocidalo, M.-A.; Leger, C.-L. α-Tocopherol Inhibits the Respiratory Burst in Human Monocytes. J. Biol. Chem. 1998, 273, 32801–32805.
- Munteanu, A.; Taddei, M.; Tamburini, I.; Bergamini, E.; Azzi, A.; Zingg, J.-M. Antagonistic Effects of Oxidized Low Density Lipoprotein and α-Tocopherol on CD36 Scavenger Receptor Expression in Monocytes. J. Biol. Chem. 2006, 281, 6489–6497.
- Zingg, J.-M.; Libinaki, R.; Meydani, M.; Azzi, A. Modulation of phosphorylation of tocopherol and phosphatidylinositol by hTAP1/SEC14L2-mediated lipid exchange. PLoS ONE 2014, 9, e101550.
- Sampath, H.; Ntambi, J.M. Polyunsaturated Fatty Acid Regulation of Genes of Lipid Metabolism. Annu. Rev. Nutr. 2005, 25, 317–340.
- Negis, Y.; Zingg, J.M.; Ogru, E.; Gianello, R.; Libinaki, R.; Azzi, A. On the existence of cellular tocopheryl phosphate, its synthesis, degradation and cellular roles: A hypothesis. IUBMB Life 2005, 57, 23–25.
- Zingg, J.-M.; Libinaki, R.; Lai, C.-Q.; Meydani, M.; Gianello, R.; Ogru, E.; Azzi, A. Modulation of gene expression by α-tocopherol and α-tocopheryl phosphate in THP-1 monocytes. Free Radic. Biol. Med. 2010, 49, 1989–2000.
- Zingg, J.-M.; Azzi, A.; Meydani, M. Induction of VEGF Expression by Alpha-Tocopherol and Alpha-Tocopheryl Phosphate via PI3Kγ/PKB and hTAP1/SEC14L2-Mediated Lipid Exchange. J. Cell. Biochem. 2015, 116, 398–407.
- Sluimer, J.C.; Gasc, J.-M.; van Wanroij, J.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjens, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, Hypoxia-Inducible Transcription Factor, and Macrophages in Human Atherosclerotic Plaques Are Correlated with Intraplaque Angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265.
- Zhu, R.; Hu, Y.; Tang, L. Reduced cardiac function and risk of venous thromboembolism in Asian countries. Thromb. J. 2017, 15, 12.
- Glynn, R.J.; Ridker, P.M.; Goldhaber, S.Z.; Zee, R.Y.L.; Buring, J.E. Effects of Random Allocation to Vitamin E Supplementation on the Occurrence of Venous Thromboembolism. Circulation 2007, 116, 1497–1503.
- Murohara, T.; Ikeda, H.; Otsuka, Y.; Aoki, M.; Haramaki, N.; Katoh, A.; Takajo, Y.; Imaizumi, T. Inhibition of Platelet Adherence to Mononuclear Cells by α-Tocopherol. Circulation 2004, 110, 141–148.
- Martin, A.; Foxall, T.; Blumberg, J.B.; Meydani, M. Vitamin E Inhibits Low-Density Lipoprotein Induced Adhesion of Monocytes to Human Aortic Endothelial Cells In Vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 429–436.
- Islam, K.N.; Devaraj, S.; Jialal, I. α-Tocopherol Enrichment of Monocytes Decreases Agonist-Induced Adhesion to Human Endothelial Cells. Circulation 1998, 98, 2255–2261.

