1. Antioxidant Mechanism
Clinical trials reported during the 1990s and early 2000s are reviewed here to gain better insights into the aetiology of vitamin E on the human vascular system that were established after its discovery.
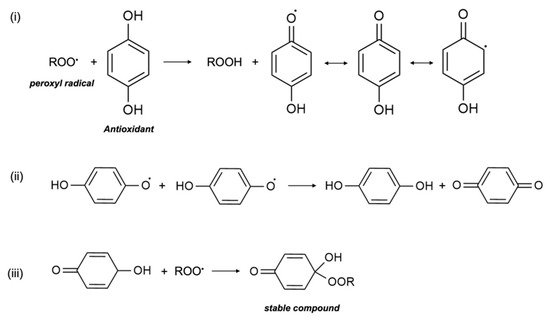
In in vitro research, vitamin E was shown to act as a scavenger of the peroxyl radical (LOO•), which helps to inhibit free-radical propagation and thereby lipid oxidation [
1]. In cells, vitamin E is found in the phospholipid bilayer of the cell membrane and in plasma lipoproteins, and protects polyunsaturated fatty acids (PUFA) in situ from oxidation by peroxyl radicals. During the process of lipid peroxidation, molecular oxygen reacts with lipid carbon-centred radicals (L•) of various organic molecules such as fatty acids, cholesterol, phospholipids, etc., to form LOO•, which then reacts with PUFA to form lipid hydroperoxide (LOOH) and another L•. Antioxidants, including vitamin E, scavenge free radicals and break the lipid peroxidation chain reaction. In the presence of LOO•, the phenolic hydroxyl group of αT undergoes an event of propagation reaction and forms into a tocopheroxyl radical, leaving LOOH and preventing the development of L• (
Figure 1). The tocopheroxyl radical is much more stable and harmless as compared to the peroxyl radical, and potentially converts into tocopherol via reduction from other antioxidants, such as ascorbic acid (vitamin C) or with another tocopheroxyl radical [
22]. Vitamin E is an extremely efficient antioxidant since its rate of reaction and scavenging of LOO• is much higher than the rate of peroxidation of lipid molecules by peroxyl radicals [
23]. In some cases, vitamin E can also act as a pro-oxidant and oxidize other lipids. Nonetheless, to date, the extent of vitamin E recycling and the conditions required for its antioxidant effect are not clearly known [
5].
Figure 1. Termination of free radicals by antioxidants. The peroxyl radical is scavenged by antioxidants e.g., α-tocopherol. The resulting metabolite either (i) stabilizes itself, (ii) combines with another similar metabolite, or (iii) scavenges another peroxyl radical.
The oxidative-modification hypothesis of atherosclerosis postulates that the accumulation of LDL in the sub-endothelium of arteries and its subsequent oxidation is the main cause of atherosclerosis. LDL accumulation initiates the recruitment of monocytes and macrophages, which further favors the peroxidation of LDL. The oxidized LDL generated is internalized by macrophages, which converts them into foam cells. Foam cells can accumulate in the sub-endothelium of arteries, which results in inflammation and plaque formation. The uptake of oxidized LDL by the scavenger–receptor pathway is not negatively regulated and leads to increased regulation of cholesterol. Chronic inflammation of endothelial cells and the narrowed lumen of arteries lead to the development of atherogenesis. Atherosclerotic arteries are a major developmental factor for various CVDs, since it narrows the lumen of arteries, cause hardening of the sub-endothelium, reducing blood flow and damaging endothelial cells. Furthermore, foam cells are cytotoxic in vascular cells, which leads to lipid and lysosomal enzyme release and vascular lesion formation. LDL oxidation and its cytotoxicity may be prevented by the incorporation of vitamin E, thus preventing the development of atherosclerosis, and subsequently reducing the risk of CVDs [
24,
25].
Various clinical trials conducted in the late 1980s and 1990s perceived vitamin E to be a strong antioxidant that aided in preventing various CVDs. Followed by the hypothesis of αT’s antioxidant role in preventing LDL oxidation, several studies conducted large-scale human trials to investigate the reduction of various CVD risks. In 1993, separate studies (the Health Professional’s Follow-Up Study and the Nurses’ Health Study) were conducted in males and females to understand the relationship between vitamin E consumption and the risk of coronary artery disease (CAD). These trials confirmed that high oral doses of both dietary and supplemental (100–250 IU/day) vitamin E reduced the risk of major coronary diseases such as CAD, non-fatal myocardial infarction (MI), and cardiovascular death by 35–40%. These studies correlated the results with the reduction of cell-meditated oxidation of LDL by vitamin E and found reduced rates of atherosclerosis and restenosis [
26,
27]. In 1995, the Cholesterol Lowering Atherosclerosis Study conducted a subgroup analysis on the supplementary intake of vitamin E and the rate of progression of CAD. Researchers conducted angiography on the subjects after 2 years of intervention and found that supplementation of >100 IU/day of vitamin E caused the least progression in coronary artery lesions due to a lower rate of stenosis [
28]. The Cambridge Heart Antioxidant Study (CHAOS) conducted in 1996 had similar angiographic findings. αT treatments were given to 2002 patients suffering from atherosclerosis for a median time of 510 days and showed a lower rate of non-fatal MI due to the reduced rate of macrophage-rich atherosclerotic lesion formation [
29]. In vitro studies also showed a direct correlation between αT levels and oxidized LDL in the prevention of arterial dysfunction; high tissue levels of αT lowered the risk of endothelial dysfunction from oxidized LDL [
30]. Another trial, known as the Secondary Prevention with Antioxidants of CVD in Endstage Renal Disease (SPACE) Study (2000), indicated that chronic hemodialysis patients have high vascular disease related mortality rates. Patients that undergo hemodialysis to treat renal diseases have elevated oxidative stress; however, vitamin E supplementation potentially prevented vascular complications, especially CVD. This observation suggests that vitamin E lower the rate of oxidized LDL production, which subsequently reduced the rates of non-fatal MI by 70%, cardiovascular mortality by 46%, and ischemic stroke by 14% [
31]. A relatively recent clinical trial on the effect of vitamins on cardiovascular health in a Russian population showed that multivitamin supplementation, including αT, had an inverse relationship with the risk of CVDs. The Russian population was characterized by a high alcohol intake, smoking, and a low intake of natural vitamin sources such as fruits and vegetables, leading to poor antioxidant status. After continuous supplementation for 8 weeks, subjects showed vastly improved antioxidant levels, including αT levels, and a reduction in heart health risk biomarkers [
32]. A study conducted in 2018 also showed that vitamin E supplementation significantly improved the vascular health of patients with Hp2-2 genotypes. Hp2-2 genes code for haptoglobin and increase the risk of endothelial dysfunction, MI, and type II diabetes, and is suggested to be regulated via vitamin E at a therapeutic level [
33].
On the other hand, it has also been reported that the protective effects of tocopherol against LDL oxidation and its vascular function may not be due its antioxidant property. It was found in a long-term clinical trial that vitamin E may not have protective effects against CVD. One important study was the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Trial (ATBC) conducted in 1996 on Finnish smokers. The effect of vitamin E on the prevention of angina pectoris was deemed unlikely to be of public health significance. Similar findings were shown in patients with intermittent claudication and those undergoing coronary angioplasty [
34]. The Heart Outcomes Prevention Evaluation—The Ongoing Outcomes (HOPE-TOO) Study additionally reported that there was no significant difference in the incidence of major cardiovascular events after daily long-term administration of a high dose (400 IU) of natural vitamin E. In fact, it further suggested an increased risk of heart failure [
35]. Similarly, the Women’s Health Study (WHS) conducted from 1992–2004 to test if vitamin E supplementation reduced the risk of CVD in women further showed a non-significant risk reduction after long-term administration of vitamin E supplementation [
36]. The Physicians’ Health Study II conducted in 2008 among middle-aged and older men did not find any correlation between supplemental use of vitamin E and the risk reduction of non-fatal MI, angina, or heart failure [
37]. This has also been supported by older studies conducted on a small-scale or population-subset level. Moreover, the MRC/BHF Heart Protection Study conceded that supplemental doses of vitamin E, along with other antioxidant vitamins, did not decrease the risk of incidence or mortality of CVD and cancer [
38]. A recent study conducted on a Singaporean population contends that vitamin E supplementation did not have any preferential benefit in patients with the Hp2-2 genotype, but rather showed an increased risk of arterial stiffness in patients with high haptoglobin concentrations [
39].
Due to the opposing views on the antioxidant effect of vitamin E in preventing various CVDs, it is recommended that more advanced clinical trials with larger test groups of participants should be investigated, with higher control on external factors such as age, eating patterns, environmental influences, etc. Additionally, a more quantitative assessment is needed to measure the oxidative stress of the human body to correctly prove the oxidation hypothesis. Moreover, the LDL oxidation rate in humans can be very slow, and atherosclerotic lesions can take many years to form. It is plausible that only a sub-population may respond to vitamin E supplementation, while some may be influenced by other environmental and genetic factors; for example, a family history of hypercholesterolemia [
25]. Alternatively, an examination of the molecular roles of vitamin E in the prevention of cardiovascular complications could lead to new insights in its functions.
2. Potential Molecular Mechanisms
While vitamin E’s antioxidant mechanism is extremely well-known and popularised in medicinal research, its bioactive effects are relatively lesser known. Research on cellular regulation by vitamin E and its involvement in metabolic pathways is new and emerging. Current studies on vitamin E agree that its antioxidant effect may not be enough to justify all of its effects on the vascular system. Nevertheless, vitamin E is also able to regulate gene expression and take part in signal transduction, enzymatic activities, and membrane phenomena of lipid fluxes. Several of these molecular mechanisms, as discussed below, suggest that the protective function of vitamin E against CVD is not merely due to its free radical scavenging property [
40].
Vitamin E protects plasma membranes from free radical damage by acting as an antioxidant. However, it also protects them and promotes their repair via modulation of the membrane by a stabilization process. For instance, the chromanol head of αT binds with phospholipids to reduce mobility and fluidity in the interior layer [
41]. Vitamin E is also able to stabilize the membrane of red blood cells and prevent hemolysis induced by vitamin A [
23]. Furthermore, it indirectly protects membrane stability by suppressing ROS formation caused by oxidized LDL, which will be discussed in the following sections.
Vitamin E can directly influence phospholipid metabolism by activating phospholipase A
2 (PLA
2), which is involved in signal transduction, and regulate the levels of lysophosphatidylcholine species (lysoPC) [
42]. LysoPC are pro-inflammatory and induce platelet activation and endothelial dysfunction in the vascular system. They form complexes with vitamin E, and thus stabilize the endothelial cells. In addition, patients with obesity and type 2 diabetes fall in the CVD risk group, where high LysoPC levels in plasma were found in these individuals. It was also shown that vitamin E forms complexes with LysoPC and prevents PLA
2 hydrolysis in rat liver lysosomal membranes [
43,
44].
In vitro studies have shown that vitamin E is able to prevent apoptosis caused by the potential cytotoxic effect of docosahexaenoic acid (DHA) in cultured cells by modulating the gene expression of the pregnane X receptor (PXR) [
45] and similar heterodimeric nuclear receptors. The positive synergy between DHA and vitamin E can also affect the regulation of DP-glucuronosyltransferase 1A1 (UGT1A1) mRNA in cell detoxification and increase stearoyl-CoA desaturase (SCD) levels [
46], which play a role in lipid biosynthesis. Moreover, αT decreases the production of caspase-3 in the caspase cascade, which guides the downregulation of oxidized LDL and ROS-activated apoptotic pathways in endothelial cells [
47].
A study showed that vitamin E induces the expression of endogenous antioxidant enzymes that prevents lipid peroxidation (and hence foam cell formation). These enzymes, namely Cu/Zn superoxide dismutase (SOD) and catalase (SOC), activate PPARγ and NF-κB redox-sensitive gene regulators [
48] in vascular cells. Additionally, vitamin E also decreases the release of inflammatory cytokines IL-1β, IL-8, and IL-6 and thus reduces inflammation [
49] developed during excessive macrophage recruitment. Moreover, vitamin E supplementation has been shown to downregulate the expression of CD36 scavenger receptors that are responsible for the uptake of cholesterol, and consequently prevent foam cell formation [
50]. Vitamin E-deficient cells also had high levels of CD36. Vitamin E upregulates the expression of PPARγ, LXRα, and ABCA1 pathways, and forms a transduction pathway for the transport and efflux of cholesterol. This results in cholesterol efflux from macrophages and the subsequent prevention of foam cell formation. Furthermore, all genes are induced in the presence of concentrated oxidized LDL, showing that vitamin E interacts with LDL via the modification of gene expression in addition to the antioxidant pathway. It can also be inferred that vitamin E supplementation can prevent the onset of hypercholesterolemia, which contributes to the onset of early atherosclerosis [
51].
The proliferation of vascular (specifically aortic) smooth muscle cells (VSMC) is associated with the risk of vascular diseases, namely atherosclerotic lesion formation, hypercholesterolemia, and hypertension [
52]. Studies have shown that αT can block VSMC proliferation via the inhibition of the isoenzyme protein kinase C (PKC) family. PKC upregulates the pathway that converts quiescent VSMC to their proliferative state. In general, PKC activates an important cell transduction pathway responsible for cell growth, proliferation, differentiation, and secretion [
53]. In addition, PKC is one of the main receptors for tumour-promoting phorbol esters. αT is a specific PKC inhibitor and prevents VSMC proliferation in several ways. Experiments have shown that αT can increase phorbol ester binding to PKC. However, the overall effect of vitamin E is preventative, and phorbol-binding does not increase PKC production in cells. It is postulated that αT inhibits phosphorylation of the 80-kDa protein, which is a PKC activation marker and enzyme substrate for the signal transduction pathway. αT also prevents the translocation of phorbol-bound PKC to the membrane after its activation, and thus reduces its cellular distribution [
52]. At the molecular level, vitamin E analogues prevent the activation of PKC by binding differentially to its diacylglycerol (DAG) binding sites [
7]. In diabetic patients (which present a high risk of developing CVD), PKC is activated by increased DAG production. However, αT is able to inhibit PKC release by stimulating the production of DAG kinase, which phosphorylates DAG [
54], and consequently lowers its levels.
Macrophages also produce tumour necrosis factor-α (TNF-α), which leads to ROS production and the inhibition of connective tissue growth factor (CTGF). CTGF is necessary in VSMC for wound repair, tissue growth, and plaque stabilization during atherosclerotic lesion formation. αT is antagonistic to TNF-α [
55] and helps in regressing CTGF downregulation induced by it [
56], and subsequently prevents ROS production, inflammation, and atherosclerotic plaque build-up. Studies have also shown that oxidized LDL causes inactivation of the endothelial derived relaxation factor (EDRF) in VSMC, which is a major risk factor for CVD; this indicates EDRF release to be indirectly regulated by αT. Oxidized LDL also degrades endothelial nitric oxide (NO), which is important for vascular health and the prevention of platelet aggregation [
57]. It stimulates the release of PKC through phorbol 12-myristate 13-acetate (PMA). PMA activates the G-protein, and the subsequent PKC release prevents endothelium-dependent arterial relaxation and receptor-mediated stimulation of NO production by the endothelium. The vascular incorporation of αT blocks PKC activation, and thus protects the endothelium from degeneration caused by oxidized LDL. This also provides further proof of vitamin E’s function against injury from oxidized LDL in endothelium tissue, as opposed to its antioxidant activity [
30]. Further investigation showed that vitamin E prevented LDL oxidation via the release of superoxide anions (O
2−) by monocytes. The enzyme NADPH oxidase is responsible for O
2− production by phagocytes. The phosphorylation of cytosolic proteins p47
phox by PKC and PMA is responsible for NADPH oxidase activation. However, αT blocked the translocation and phosphorylation of p47
phox by PMA and PKC, inhibited O
2− production by the monocytes [
50,
58], and consequently prevented the oxidation of LDL.
Furthermore, oxidized LDL also activates enzyme protein kinase B (PKB/Akt) in VSMC. PKB/Akt is a major enzyme responsible for controlling various cellular processes such as cell migration, cell apoptosis, gene expression, lipid metabolism, free radical production, etc. [
7]. During atherosclerotic plaque formation, increased PKB/Akt levels have been observed. Vitamin E inhibits the phosphorylation of Ser473, the key regulatory site of PKB/Akt, and subsequently regulates the production of the PKB enzyme (independent of PKC regulation). Moreover, vitamin E decreases oxidized LDL uptake by THP-1 macrophages by preventing oxidized LDL-induced Ser473 phosphorylation. This further downregulates CD36 scavenger receptors via PPAR in the ox-LDL/CD36/PKB/PPARγ pathway. As a result, it decreases overall lipid uptake and biosynthesis in cells [
59]. Cholesterol biosynthesis and lipid exchange is also prevented by the binding of αT to tocopherol-associated proteins (TAP 1/2/3), which inhibit enzymes squalene epoxidase and HMG-CoA reductase [
60].
Vitamin E has been established as an antioxidant that protects monounsaturated fatty acids and polyunsaturated fatty acids (MUFA/PUFA). However, it can also indirectly regulate the gene expression and production of lipid mediators regulated by MUFA/PUFA, which control membrane receptors, membrane lipid composition, signal transduction enzymes, transcription factors (including PPARγ, hepatocyte nuclear factor HNF-4α, NRF2, LXRα/β, etc.), and lipid metabolism enzymes [
61]. These play a significant role in regulating the metabolism of cholesterol, carbohydrates, and triglycerides in cells [
7] and ultimately determine the risk of atherosclerosis, hyperlipidemia, and hypercholesterolemia.
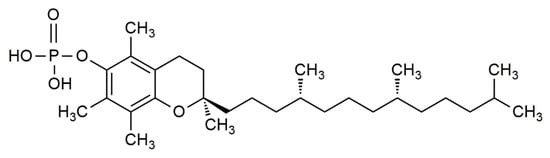
Newer studies have further laid an emphasis on the molecular effect of α-tocopherol phosphate (αTP) (
Figure 2) in modulating gene expression. αTP is a natural analogue of αT that is found extensively in foods and in the human body. While αTP has no known antioxidant effect, it is shown to act as a lipid mediator in membranes, as a cofactor for enzymes, and as a receptor for various transcription factors for mRNA [
62]. αTP is shown to be more potent than αT in reducing the expression of CD36 scavenger receptors and the proliferation of human THP-1 monocytes, thus lowering inflammation and atherosclerotic lesion formation. Additionally, αTP stimulates human TAP-1, which consequently interacts with the phosphatidylinositol-3-kinase (PI3K/Akt) signal transduction pathway to modulate the expression of several vascular endothelial growth factor (VEGF) genes [
60,
63,
64]. It is shown that stimulation of VEGF genes by αTP is essential for cell and wound repair, tissue remodelling, and stimulates vascular permeability, vasculogenesis, and angiogenesis, which can prevent hypoxia in atherosclerotic regions at an early stage [
63,
65]. Moreover, VEGF expression can be further promoted by the production of αTP in VSMC [
60].
Figure 2. The chemical structure of α-tocopherol phosphate (αTP).
CVD includes a range of disorders and conditions, with the most serious ones being CAD and stroke. Venous thromboembolism (VTE) is the third most common CVD after these two and refers to the formation of blood clotting in the deep veins of the limbic and groin region; when the clot breaks, it is circulated in the blood stream and transported to the lungs, leading to a primary lung embolism. In extreme cases, it can also cause right heart failure [
66]. A clinical trial conducted on women showed that a supplemental dose of vitamin E can reduce the hazard of VTE significantly by 21%, and the hazard of unprovoked VTE by 27%. Furthermore, the risk of a primary embolism was reduced by 28%. The hypothetical reason behind this is the anticoagulation effect of vitamin E (when vitamin K levels in the body are low) as well as the inhibition of platelet adherence. Hence, it can be potentially used to prevent first or recurrent VTEs in the body [
67]. A deeper look into the antithrombotic effects of vitamin E suggests that αT can prevent thrombus formation via the inhibition of platelet adherence to mononuclear cells (MNC). Oral supplementation of vitamin E given to healthy subjects suppressed PMA-mediated P-selectin expression, interactions between platelets and MNCs, platelet PKC activity, and overall agonist-induced platelet aggregation ex vivo. Since P-selectin is prothrombotic, and PKC promotes platelet-MNC interaction, vitamin E can have potential uses in the treatment of thrombosis, which reduces the risk of atherosclerosis and CAD. This also highlights the differing effects of vitamin E in arterial and venous events [
68]. Further evidence showed that supplemental vitamin E downregulates gene expression for the intercellular cell adhesion molecule (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which are induced in the presence of elevated oxidized LDL [
69], and subsequently reduces the adhesive capabilities of cellular blood components to endothelium lining of the blood vessels. In vitro studies on human cell lines also showed that αT downregulates the expression of adhesion molecules CD11b and very late antigen-4 (VLA-4), which in turn suppress the migration and adhesion of activated monocytes [
70] to the endothelium. Altogether, the downregulation of ICAM-1, VCAM-1, CD11b, and VLA-4 by αT directly reduced the risk of inflammation and thrombosis in the vascular system.