Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antiopi Vardaxi | + 2654 word(s) | 2654 | 2022-02-22 07:12:52 | | | |
| 2 | Rita Xu | Meta information modification | 2654 | 2022-02-24 03:27:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vardaxi, A. Polymeric Nanostructures Containing Proteins. Encyclopedia. Available online: https://encyclopedia.pub/entry/19827 (accessed on 24 June 2026).
Vardaxi A. Polymeric Nanostructures Containing Proteins. Encyclopedia. Available at: https://encyclopedia.pub/entry/19827. Accessed June 24, 2026.
Vardaxi, Antiopi. "Polymeric Nanostructures Containing Proteins" Encyclopedia, https://encyclopedia.pub/entry/19827 (accessed June 24, 2026).
Vardaxi, A. (2022, February 23). Polymeric Nanostructures Containing Proteins. In Encyclopedia. https://encyclopedia.pub/entry/19827
Vardaxi, Antiopi. "Polymeric Nanostructures Containing Proteins." Encyclopedia. Web. 23 February, 2022.
Copy Citation
Over the last three decades, proteins and peptides have attracted great interest as drugs of choice for combating a broad spectrum of diseases, including diabetes mellitus, cancer, and infectious and neurological diseases. However, the delivery of therapeutic proteins to target sites should take into account the obstacles and limitations related to their intrinsic sensitivity to different environmental conditions, fragile tertiary structures, and short half-life. Polymeric nanostructures have emerged as competent vehicles for protein delivery, as they are multifunctional and can be tailored according to their peculiarities.
polymeric nanostructures
therapeutic proteins
protein delivery
amphiphilic block copolymers
1. Introduction
Since human insulin was approved by the FDA (Food and Drug Administration) in 1982, when it became the first commercially available recombinant therapeutic protein [1], several therapeutic proteins and peptides have been endorsed for clinical use and others are under development for the therapy of various diseases such as cancer [2], hepatitis [3], and diabetes [4][5]. Currently, 40% of the 6000 biopharmaceutical products that are in advanced stages of clinical development are protein-based, indicating the prevalence of these products in the future [6].
Proteins and peptides are the dominant biological macromolecules of life that perform essential biochemical functions inside cells, such as enzyme catalysis and signal transduction, as well as being involved in many pathological conditions, such as diabetes and cancer [7]. Cancer onset occurs with the mutation of one or more genes, which causes either the formation of an aberrant protein or hinders the generation of a protein. Irregular proteins deliver dissimilar information to ordinary proteins. This provokes the intractable multiplication of cells, which eventually become cancerous [8]. Both proteins and peptides are comprised of amino acid units and are held together by peptide bonds. Generally, proteins consist of 50 or more amino acids, while peptides are distinguished from proteins based on their size and structure, because they are made up of between 2 and 50 amino acids. Furthermore, peptides are divided into oligopeptides and polypeptides, with the first one consisting of 2–20 amino acids and the last one consisting of 50 or more amino acids [9]. The structure of a protein can be classified into four different forms, namely the primary, secondary, tertiary, and quaternary forms, while the entire three-dimensional structure is dependent on the folding of polyamino acid chains (Scheme 1). The primary structure is related to the sequence of 20 common amino acids located in the protein structure, whereas the more complex structures resulting from the folding and interactions between amino acids are the secondary, tertiary, and quaternary forms [7].
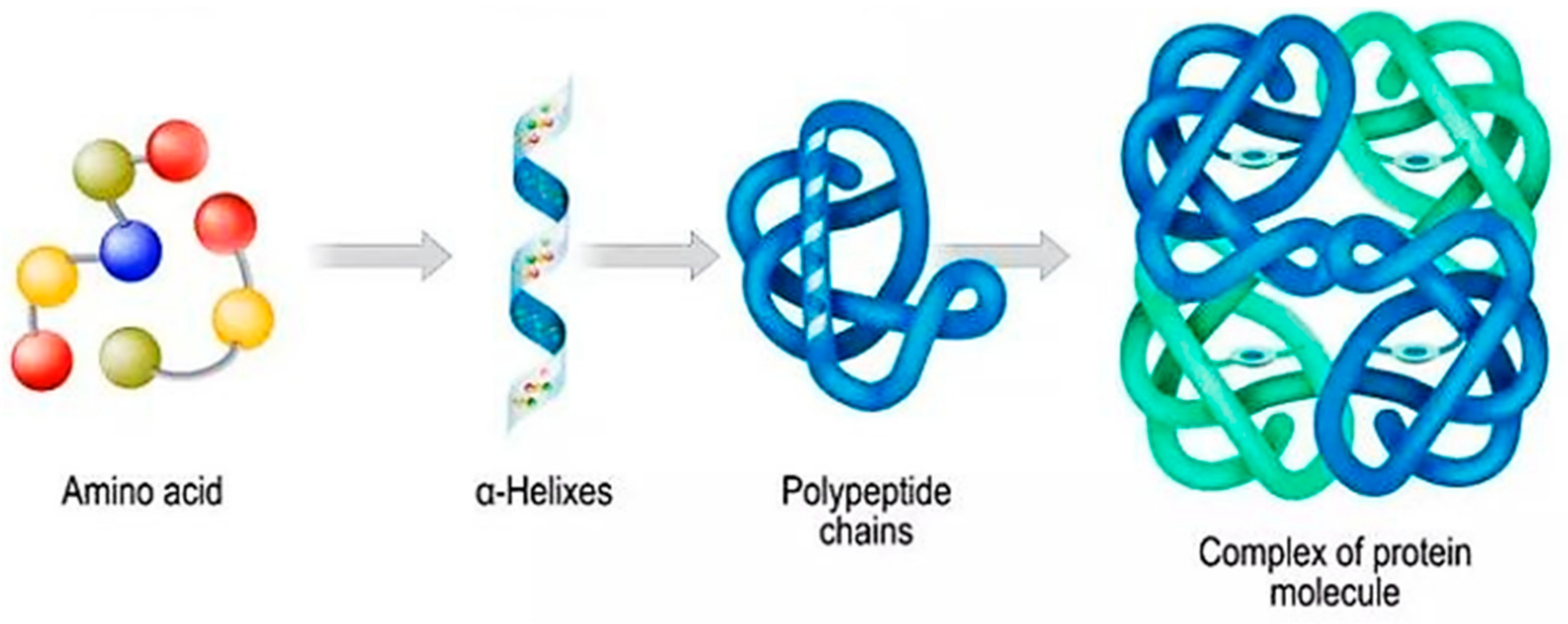
Scheme 1. Structural classification of proteins [10].
Therapeutic proteins and peptides exhibit tremendous advantages compared to conventional small-molecule drugs. They have advanced specificity and better activity; thus, they cannot be simulated by other chemical compounds. Additionally, they possess less toxicity, which is associated with an immune response during administration [9][11]. However, the high molecular weight of proteins and peptides in conjunction with their varying surface charges and fragile tertiary structures contribute to their limitations in their use as drug molecules, as the capability of their stand-alone delivery into the intracellular space is limited [12]. Furthermore, their high hydrophilicity and the presence of charged groups subsequently lead to poor cell membrane permeability. Moreover, proteins and peptides are susceptible to different environmental conditions, including ionic strength and pH or temperature variations, resulting in the alteration of their physical stability. For instance, their charged groups are vulnerable to the surrounding water molecules because they can interact with them and produce hydrogen bonds, while the peptide bonds in their structure can be influenced by the proteolytic enzymes, which cause proteolytic hydrolysis. Additionally, the restricted permeability across gastrointestinal mucosa, the degradation by enzymes in the gastrointestinal tract (GIT), the reticuloendothelial system (RES) clearance, and the elimination during first-pass clearance effects are obstacles of paramount importance during either oral or parenteral administration of therapeutic proteins and peptides, thereby limiting their clinical application [7][12][13][14][15]. Therefore, several strategies, including the coating of proteins with a protective polymeric layer or their delivery through vehicles at the nanosize scale, are being investigated in order to overcome the aforementioned limitations [16][17].
Regarding the complexity of therapeutic protein and peptide delivery, the field of nanotechnology has effectively bridged the gap, as it provides several nanoscale systems that have the required encapsulating properties, further offering a better pharmacokinetic profile [18][19]. Such nanosystems include lipid nanoparticles [20], liposomes [21], polymeric nanoparticles [22], and magnetic nanoparticles [23], among others. To date, polymeric nanocarriers have gained significant interest as protein delivery vehicles thanks to their multifunctionality. Thus, the enhanced biocompatibility and bioavailability enable these structures to be compatible with living tissues without causing toxic effects in the host, while at the same time protecting the cargo from extreme environmental conditions, subsequently permitting targeted delivery [12][24]. Block copolymers have been extensively utilized in nanostructure construction owing to their structural adaptability, chemical composition, and flexibility. Moreover, the variable molecular weight (Mw) and tunable chemical properties can be tailored to the requirements of each protein–block copolymer [25]. Furthermore, the amphiphilic block copolymers (AmBCs), which exhibit the ubiquitous feature of self-assembly in selective solvents, form structures such as micelles or polymersomes that are eligible vehicles for protein delivery [26][27]. Lastly, polyelectrolytes, amphiphilic block copolymers (AmBCs) comprising at least one polyelectrolyte block, and surfactants belong to an interesting field of macromolecules for clinical use [28].
2. Polymeric Nanostructures
Polymeric materials have opened new horizons in protein and drug delivery; hence, they have been adopted as subjects of great interest from scientists working in these fields. Their high biodegradability, the adjustable control of both the structure and chemical arrangement, their colloidal stability, and the potential for integration of a plethora of either hydrophobic or hydrophilic molecules are a few of the promising properties that they possess [29][30]. Nevertheless, it is necessary for these nanostructures not to trigger cytotoxicity causing toxic impacts to healthy tissues, but to provide proper host response and extended therapeutic effects [31]. Moreover, these novel multifunctional nanoplatforms can contribute to diagnosis and therapy (theranostics) in a single system [32][33].
Polymeric nanostructures are formed through the self-assembly process of short polymer chains or more complicated polymer architectures, such as dendrimers and multibranched polymers. Stimuli-responsive polymers, so-called smart polymers, are the key factor in the latest generation of supramolecular nanocarriers, which are formed through the combination of the two categories. These polymers are able to undergo physicochemical changes or rearrangements due to their response to a specific stimulus, whether that be endogenous, such as to pH and redox variations, hypoxia, and enzymes, or exogenous, such as temperature alterations, light exposure, magnetic fields, and ultrasound, thereby enabling controlled release of active pharmaceutical ingredients (API) at specific sites [32][34][35].
Block copolymers (BCPs) with precise molecular weights, compositions, and architectures have received massive attention thanks to their ability to form explicit structures at the nanoscale that can be used as nanocarriers of biomolecules [36]. They are comprised of two, three, or more distinct blocks of diverse chemical groups, joined together with covalent bonds, leading to the construction of diblock, triblock, or multiblock copolymers, respectively. Each block is composed of the repetition of a single monomeric unit. Furthermore, BCPs are classified along with their structural array to linear, star-shaped, graft, branched, and cyclic architectures, whereas their ultimate configuration arises from the regulation of parameters in the synthetic process (Scheme 2). The Flory–Huggins interaction parameter, the polymerization degree, the volume fraction, the molecular weight, and the proportion of each component block result in the differential demonstration of nanomorphologies, e.g., spherical, cylindrical, lamellar [37][38].
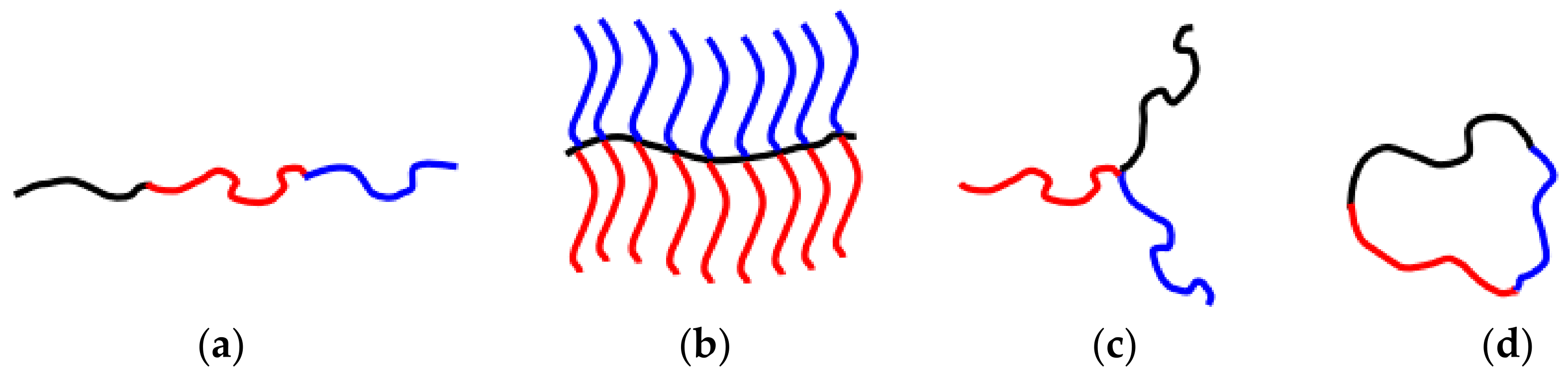
Scheme 2. Representative examples of (a) linear triblock terpolymer, (b) “comb” graft polymers, (c) “miktoarm” star terpolymers, and (d) cyclic block terpolymers [38].
Living or controlled radical polymerization (CRP) strategies are the most facile and multifaceted path for BC preparation. They are congruent with an abundance of monomers. Furthermore, they exhibit sufficient acceptance of functional groups. Three main controlled radical polymerization (CRP) methodologies have been reported regarding the preparation of BCs, including atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT) radical polymerization, and nitroxide-mediated polymerization (NMP) [39].
The RAFT approach has been established as the most advantageous method for acquiring well-defined BCs, as it is an easily manipulable method that is compatible with a wide range of monomers that bear functional segments. In addition, the RAFT protocol allows control over molecular characteristics such as the molecular weight and composition through the proper adaptation of the polymerization conditions. The diverse molecular architectures, absence of catalysts, possible realization of the polymerization process in aqueous media, soothing experimental conditions, high precision over the final product with well-appointed side-groups, and post-polymerization functionalization are only a few of the beneficial features that RAFT polymerization provides [39][40][41].
Block copolymers can be further classified according to the nature of the constituent blocks and their affinity with water. AmBCs consisting of at least one hydrophobic and one hydrophilic block self-assemble in aqueous solutions, further forming micellar-like nanostructures or nanoaggregates. The self-assembly process of a block copolymer results in the formation of a corona–shell-type micelle, whereby the hydrophobic block comprises the core of the nanoparticle and the hydrophilic block comprises the shell that surrounds the core and prevents the contact of the hydrophobic part with water molecules [5][26][42]. During the self-assembly process, electrostatic interactions, hydrogen bonding, π–π aromatic stacking, and van der Waals forces altogether influence the organization of macromolecules, while by tailoring the experimental parameters, the preferable structures can be collected. This procedure also promotes reduced interfacial free energy, which arises from the need to decrease the interfacial area of non-soluble parts. Moreover, according to solvophilic and solvophobic segments of AmBCs, distinguishable morphologies such as micelles, lamellae, and vesicles can be observed. Remarkably, AmBCs micelles contribute to the delivery of solubilized water-insoluble molecules such as proteins, as reviewed in the following section [37][43]. Furthermore, the amenability of molecular features of the self-assembly process provides the ability of different polymers to be combined. Self-assemblies of mixtures of random and block copolymers can be tuned to distribute their hydrophobic parts in a controlled way, taking advantage of the statistical distribution and covalent connection along the polymer chains. However, these blended polymeric coassemblies require further investigation in terms of the effectively distinct release of drugs and proteins [42][44][45].
Among the AmBC-based nanoassemblies, the ones obtained via the self-association of neutral copolymers are influenced by the thermodynamic equilibrium, whereas polyelectrolyte block copolymers are controlled by the strong electrostatic interactions and the release of counterions [46]. Polyelectrolytes have ionizable units, either acidic or basic, which accept or donate protons, respectively, and respond to exposure to different stimuli. In other words, polyelectrolytes can be considered as polymers constituted from repeating ionizable units. Moreover, the self-assembled polyelectrolyte block copolymers integrate the key characteristics of BCs, polyelectrolytes, and surfactants; hence, the ease of structural modification for these intriguing classes of macromolecules allows various possibilities for utilization as nanostructural delivery vehicles of drugs, proteins, peptides, and genes. These responsive nanoassemblies can deliver the therapeutic agents through electrostatic interactions, as discussed comprehensively in the following chapter [28][46][47][48].
3. Linking Polymers, Proteins, and Peptides towards the Formation of Polymer–Protein–Peptide Nanostructures
3.1. Polymer–Protein Conjugation
Protein–polymer bioconjugation comprises an approach that results in hybrid biomacromolecules that combine both the efficient and morphological features of synthetic polymers and natural proteins [49]. The process of polymer–protein conjugation is accomplished via three different, the “grafting to”, “grafting from”, and “grafting through” strategies (Scheme 3). The “grafting to” method is based on the anchorage of polymers to proteins through coupling interactions, while the polymer moiety is produced separately and prior to the final step of hybrid molecule formation. However, the low yield of the reactions resulting from the steric hindrance among these macromolecules limits its further utilization. Moving on, the “grafting from” approach was demonstrated when living polymerizations techniques came to the forefront and entails the in situ growth of a polymer chain from the protein or peptide to produce well-defined hybrid architectures. A small-molecule initiator or chain transfer agent (CTA) is primarily amended with the target protein through bioconjugation reactions, subsequently producing a macroinitiator from which controlled polymerization is generated in order for the polymer chain elongation to be completed. Among the living or controlled radical polymerization (CRP) techniques, reversible-addition−fragmentation chain transfer (RAFT) [40] and atom transfer radical polymerization (ATRP) [50] fulfill the requirements in terms of the controllable conjugation site, polydispersity, and chain length, allowing an exceptional yield of the hybrid molecule [51][52]. To conclude, the latter strategy of polymer–protein conjugation is called the “grafting through” strategy and is accomplished either after the synthesis of a macromonomer–protein complex for further polymerization or by the conjugation of multiple proteins to a polymer pursued via the polymerization and formation of a comb-like structure with high density. However, the low polymerization degree and complicated nature of the final product restricted the usage of this approach [52].
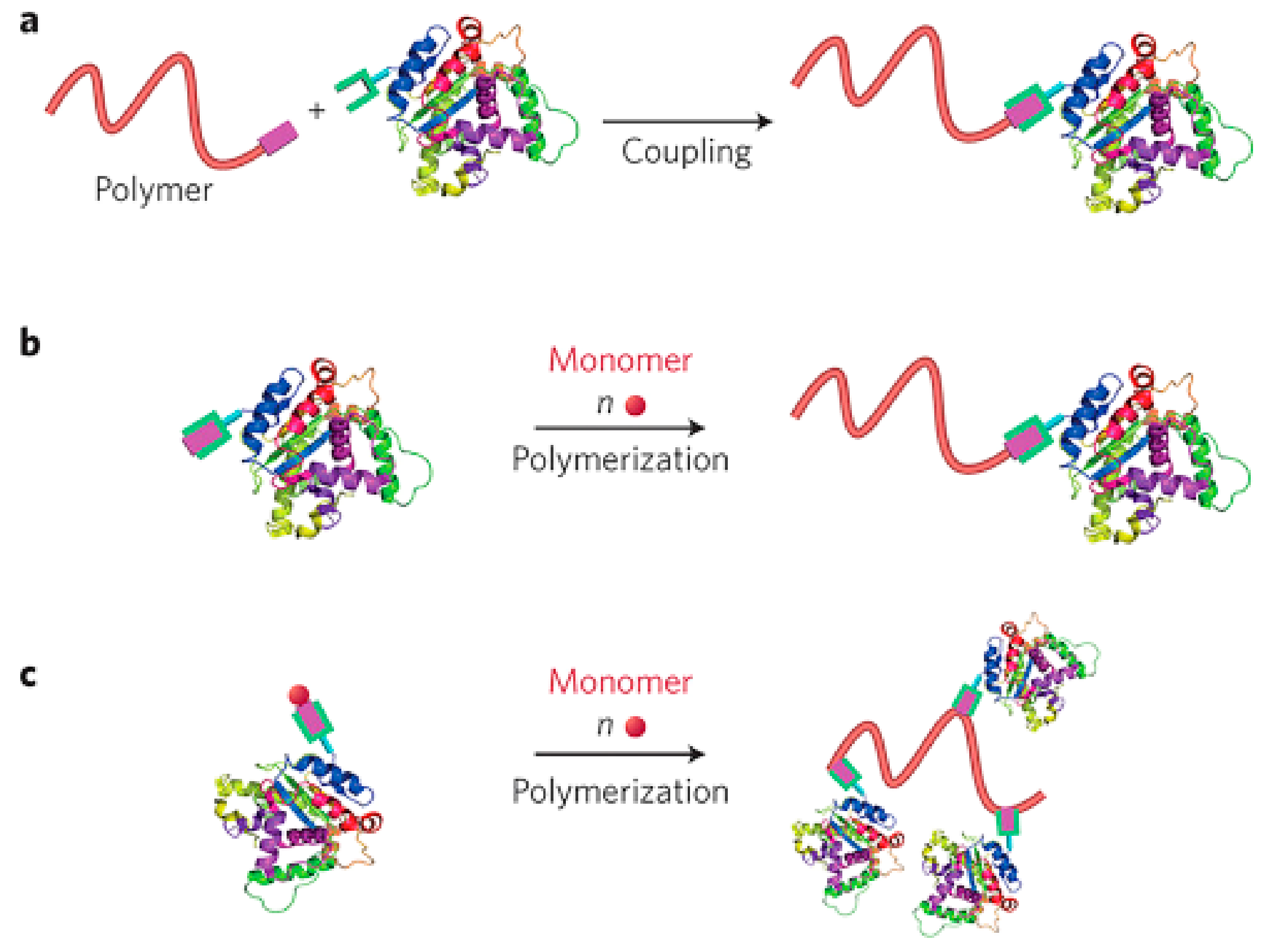
Scheme 3. (a) Grafting to, (b) Grafting from, and (c) grafting through methods used for preparing protein–polymer bioconjugates. Reprinted with permission from Ref. [52]. Copyright 2018, American Chemical Society.
3.2. Chemical Bonding of Polymer Chains with Protein
Due to the increasing interest in polymer–protein conjugates, scientists have benefited from the properties of specific polymers and applied them for protein stabilization or functionalization. Since the first covalent bonding of poly(ethylene glycol) (PEG) to bovine serum albumin (BSA) introduced by Abuchowski and colleagues in 1997 [53], numerous publications have reported on the conjugation of polymer chains to proteins. Proteins are fragile molecules that are able to be denatured when exposed to different environments and have a low half-life in blood and tissues. Hence, the chemical bonding with polymer chains increases the hydrodynamic volume while offering improved solubility, better physiological stability, and reduced immunogenicity [13][52]. The covalent conjugation with PEG polymers, also known as PEGylation, is one of the widespread strategies used for protein guarding and stabilization [54][55]. To date, over 10 PEGylated therapeutic proteins have been approved by the FDA, including Adagen®, Somavert®, Oncaspar®, and Naloxego®, which have been introduced to the pharmaceutical market, whilst other products are in clinical development [56]. Although the effective stabilization of therapeutic proteins is achieved during PEGylation, the addition of PEG chains diminishes their binding affinity and bioactivity. Zwitterionic polymers, which are polyelectrolytes characterized by equal positive and negative groups on the chains and an overall neutral charge, can be applied to overcome the weaknesses of PEG conjugation [57]. Moreover, instead of linear chains of PEG, branched or graft-like polymers such as poly(oligo(ethylene glycol) methyl ether methacrylate (POEGMA) could be alternatively utilized to improve the pharmacokinetics of therapeutic proteins. The methacrylate part in the monomer enables the site-specific polymer conjugates to be achieved using the “grafting from” strategy, while the side ethylene glycol chains demonstrate stealth properties [58].
On the contrary to the covalent-based bonding of polymer–protein bioconjugates, which generates powerful and stable connections between them, the non-covalent strategy produces architectures with superior specificity and reversibility [52]. Non-covalent approaches to biocomplexation rely on interactions such as electrostatic interactions, hydrophobic interactions, hydrogen bonding, and protein–polymer coordination, yet this linking is prone to different stimuli, such as salt, temperature, and pH; thus, the environmental alterations may influence the core–corona structures of such polymeric nanoparticles, leading to their dissociation [59]. Additionally, studies have proposed non-covalent PEGylation as a favorable tailoring approach for managing protein stability. Appropriate candidate for the non-covalent binding of PEG to protein surfaces could be diblock copolymers, including PEGylated polyelectrolytes. The attachment via strong electrostatic interactions prevents protein denaturation [60][61].
3.3. Protein Encapsulation
Proteins can be loaded into polymeric nanocarriers to provide protection from extreme environmental conditions and to extend their function until they are efficiently released at the target site [62]. Protein encapsulation can be conducted through two approaches, physical embedding and chemical bonding. Hydrophobic connections and electrostatic interactions are involved in physical embedding, whereas chemical linking of PEG (PEGylation) is involved in the chemical bonding process. Apropos of hydrophobic association, therapeutic proteins are encapsulated in the micellar core of amphiphilic block copolymers when micellization occurs [63]. On the other hand, when a block copolymer with a neutral and a charged block is mixed with an oppositely charged protein, polyion complex micelles (PIC) are formed in aqueous media with a narrow size distribution. Electrostatic interactions are mainly responsible for the PIC–protein formations, while hydrogen bonding can sufficiently contribute to the binding of the polymer and protein [64].
References
- Ye, C.; Venkatraman, S. The long-term delivery of proteins and peptides using micro/nanoparticles: Overview and perspectives. Ther. Deliv. 2019, 10, 269–272.
- Chen, J.; Zou, Y.; Deng, C.; Meng, F.; Zhang, J.; Zhong, Z. Multifunctional Click Hyaluronic Acid Nanogels for Targeted Protein Delivery and Effective Cancer Treatment in Vivo. Chem. Mater. 2016, 28, 8792–8799.
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to combat viral infectious diseases. Peptides 2020, 134, 170402.
- Washburn, R.L.; Mueller, K.; Kaur, G.; Moreno, T.; Moustaid-Moussa, N.; Ramalingam, L.; Dufour, J.M. C-Peptide as a Therapy for Type 1 Diabetes Mellitus. Biomedicines 2021, 9, 270.
- Imran, M.; Shah, M.R.; Shafiullah. Chapter 10—Amphiphilic block copolymers–based micelles for drug delivery. In Design and Development of New Nanocarriers, 1st ed.; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 365–400.
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145.
- Deb, P.K.; Al-Attraqchi, O.; Chandrasekaran, B.; Paradkar, A.; Tekade, R.K. Chapter 16—Protein/Peptide Drug Delivery Systems: Practical Considerations in Pharmaceutical Product Development. In Basic Fundamentals of Drug Delivery, 1st ed.; Tekade, R.K., Ed.; Elsevier, Academic Press: Cambridge, MA, USA, 2019; pp. 651–684.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724.
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. 2015, 20, 122–128.
- Dastider, D.; Jyoti Sen, D.; Kumar Mandal, S.; Bose, S.; Ray, S.; Mahanti, B. Hand santizers bid farewell to germs on surface area of hands. Eur. J. Pharm. Sci. 2020, 7, 648–656.
- Lagassé, H.A.D.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113.
- Zhao, H.; Lin, Z.Y.; Yildirimer, L.; Dhinakar, A.; Zhao, X.; Wu, J. Polymer-based nanoparticles for protein delivery: Design, strategies and applications. J. Mater. Chem. B 2016, 4, 4060–4071.
- Hou, Y.; Lu, H. Protein PEPylation: A New Paradigm of Protein–Polymer Conjugation. Bioconjug. Chem. 2019, 30, 1604–1616.
- Srivastava, S.; Sharma, V.; Bhushan, B.; Malviya, R.; Awasthi, R.; Kulkarni, G.T. Nanocarriers for protein and peptide delivery: Recent advances and progress. J. Res. Pharm. 2021, 25, 99–116.
- Khodabakhsh, F.; Salimian, M.; Hedayati, M.H.; Ahangari Cohan, R.; Norouzian, D. Challenges and advancements in the pharmacokinetic enhancement of therapeutic proteins. Prep. Biochem. Biotechnol. 2021, 51, 519–529.
- Dellas, N.; Liu, J.; Botham, R.C.; Huisman, G.W. Adapting protein sequences for optimized therapeutic efficacy. Curr. Opin. Chem. Biol. 2021, 64, 38–47.
- Ding, S.; Zhang, N.; Lyu, Z.; Zhu, W.; Chang, Y.-C.; Hu, X.; Du, D.; Lin, Y. Protein-based nanomaterials and nanosystems for biomedical applications: A review. Mater. Today 2021, 43, 166–184.
- Le Saux, S.; Aubert-Pouëssel, A.; Ouchait, L.; Mohamed, K.E.; Martineau, P.; Guglielmi, L.; Devoisselle, J.-M.; Legrand, P.; Chopineau, J.; Morille, M. Nanotechnologies for Intracellular Protein Delivery: Recent Progress in Inorganic and Organic Nanocarriers. Adv. Ther. 2021, 4, 2100009.
- Zeb, A.; Rana, I.; Choi, H.-I.; Lee, C.-H.; Baek, S.-W.; Lim, C.-W.; Khan, N.; Arif, S.T.; Sahar, N.U.; Alvi, A.M.; et al. Potential and Applications of Nanocarriers for Efficient Delivery of Biopharmaceuticals. Pharmaceutics 2020, 12, 1184.
- Hirai, Y.; Hirose, H.; Imanishi, M.; Asai, T.; Futaki, S. Cytosolic protein delivery using pH-responsive, charge-reversible lipid nanoparticles. Science 2021, 11, 19896.
- Jash, A.; Ubeyitogullari, A.; Rizvi, S.S.H. Liposomes for oral delivery of protein and peptide-based therapeutics: Challenges, formulation strategies, and advances. J. Mater. Chem. B 2021, 9, 4773–4792.
- Mansoor, S.; Kondiah, P.P.D.; Choonara, Y.E.; Pillay, V. Polymer-Based Nanoparticle Strategies for Insulin Delivery. Polymers 2019, 11, 1380.
- Rebekah, A.; Sivaselvam, S.; Viswanathan, C.; Prabhu, D.; Gautam, R.; Ponpandian, N. Magnetic nanoparticle-decorated graphene oxide-chitosan composite as an efficient nanocarrier for protein delivery. Colloids Surf. A Physicochem Eng. Asp. 2021, 610, 125913.
- Abasian, P.; Ghanavati, S.; Rahebi, S.; Nouri Khorasani, S.; Khalili, S. Polymeric nanocarriers in targeted drug delivery systems: A review. Polym. Adv. Technol. 2020, 31, 2939–2954.
- Agrahari, V. Advances and applications of block-copolymer-based nanoformulations. Drug Discov. 2018, 23, 1139–1151.
- Karayianni, M.; Pispas, S. Self-Assembly of Amphiphilic Block Copolymers in Selective Solvents. In Fluorescence Studies of Polymer Containing Systems, 1st ed.; Procházka, K., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 27–63.
- Perin, F.; Motta, A.; Maniglio, D. Amphiphilic copolymers in biomedical applications: Synthesis routes and property control. Mater. Sci. Eng. C 2021, 123, 111952.
- Gao, S.; Holkar, A.; Srivastava, S. Protein-Polyelectrolyte Complexes and Micellar Assemblies. Polymers 2019, 11, 1097.
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 2019, 3702518.
- Venditti, I. Morphologies and functionalities of polymeric nanocarriers as chemical tools for drug delivery: A review. J. King Saud. Univ. Sci. 2019, 31, 398–411.
- Kopeček, J.; Yang, J. Polymer nanomedicines. Adv. Drug Deliv. Rev. 2020, 156, 40–64.
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-Responsive Polymeric Nanocarriers for Drug Delivery, Imaging, and Theragnosis. Polymers 2020, 12, 1397.
- Zhang, H.; Mi, P. 12—Polymeric Micelles for Tumor Theranostics. In Theranostic Bionanomaterials, 1st ed.; Cui, W., Zhao, X., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 289–302.
- Javan Nikkhah, S.; Thompson, D. Molecular Modelling Guided Modulation of Molecular Shape and Charge for Design of Smart Self-Assembled Polymeric Drug Transporters. Pharmaceutics 2021, 13, 141.
- Lombardo, D.; Kiselev, M.A.; Magazù, S.; Calandra, P. Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Adv. Condens. Matter Phys. 2015, 2015, 151683.
- Díez-García, I.; Santamaria-Echart, A.; Eceiza, A.; Tercjak, A. Triblock copolymers containing hydrophilic PEO blocks as effective polyols for organic solvent-free waterborne poly(urethane-urea)s. React. Funct Polym. 2018, 131, 1–11.
- El Jundi, A.; Buwalda, S.J.; Bakkour, Y.; Garric, X.; Nottelet, B. Double hydrophilic block copolymers self-assemblies in biomedical applications. Adv. Colloid Interface Sci. 2020, 283, 102213.
- Feng, H.; Lu, X.; Wang, W.; Kang, N.G.; Mays, J.W. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494.
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447.
- Fairbanks, B.D.; Gunatillake, P.A.; Meagher, L. Biomedical applications of polymers derived by reversible addition—Fragmentation chain-transfer (RAFT). Adv. Drug Deliv. Rev. 2015, 91, 141–152.
- Truong, N.P.; Jones, G.R.; Bradford, K.G.E.; Konkolewicz, D.; Anastasaki, A. A comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859–869.
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892.
- Torres, J.; Dhas, N.; Longhi, M.; García, M.C. Overcoming Biological Barriers with Block Copolymers-Based Self-Assembled Nanocarriers. Recent Advances in Delivery of Anticancer Therapeutics. Front. Pharmacol. 2020, 11, 1840.
- Bodratti, A.M.; Alexandridis, P. Amphiphilic block copolymers in drug delivery: Advances in formulation structure and performance. Expert. Opin. Drug Deliv. 2018, 15, 1085–1104.
- Jiang, Z.; Liu, H.; He, H.; Ribbe, A.E.; Thayumanavan, S. Blended Assemblies of Amphiphilic Random and Block Copolymers for Tunable Encapsulation and Release of Hydrophobic Guest Molecules. Macromolecules 2020, 53, 2713–2723.
- Liu, L.-Y.; Xia, G.; Feng, Z.-J.; Hao, Q.-H.; Tan, H.-G. Self-assembly of polyelectrolyte diblock copolymers at monovalent and multivalent counterions. Soft Matter 2019, 15, 3689–3699.
- Demetzos, C. Application of Nanotechnology in Drug Delivery and Targeting. In Pharmaceutical Nanotechnology: Fundamentals and Practical Applications; Springer: Singapore, 2016; pp. 77–145.
- Jayasuriya, A.C. 8—Production of micro- and nanoscale chitosan particles for biomedical applications. In Chitosan Based Biomaterials Volume 1; Jennings, J.A., Bumgardner, J.D., Eds.; Woodhead Publishing: Sawston, Cambridge, UK, 2017; pp. 185–209.
- Theodorou, A.; Liarou, E.; Haddleton, D.M.; Stavrakaki, I.G.; Skordalidis, P.; Whitfield, R.; Anastasaki, A.; Velonia, K. Protein-polymer bioconjugates via a versatile oxygen tolerant photoinduced controlled radical polymerization approach. Nat. Commun. 2020, 11, 1486.
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, e1706441.
- Hou, W.; Wei, L.; Liu, L.; Zhao, H. Surface Coassembly of Polymer Brushes and Polymer–Protein Bioconjugates: An Efficient Approach to the Purification of Bioconjugates under Mild Conditions. Biomacromolecules 2018, 19, 4463–4471.
- Wang, Y.; Wu, C. Site-Specific Conjugation of Polymers to Proteins. Biomacromolecules 2018, 19, 1804–1825.
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581.
- Kariduraganavar, M.Y.; Heggannavar, G.B.; Amado, S.; Mitchell, G.R. Chapter 6—Protein Nanocarriers for Targeted Drug Delivery for Cancer Therapy. In Nanocarriers for Drug Delivery, 1st ed.; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 173–204.
- Rondon, A.; Mahri, S.; Morales-Yanez, F.; Dumoulin, M.; Vanbever, R. Protein Engineering Strategies for Improved Pharmacokinetics. Adv. Funct. Mater. 2021, 31, 2101633.
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864.
- Harijan, M.; Singh, M. Zwitterionic polymers in drug delivery: A review. J. Mol. Recognit. 2022, 35, e2944.
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298.
- Ju, Y.; Zhang, Y.; Zhao, H. Fabrication of Polymer–Protein Hybrids. Macromol. Rapid Commun. 2018, 39, 1700737.
- Kurinomaru, T.; Kuwada, K.; Tomita, S.; Kameda, T.; Shiraki, K. Noncovalent PEGylation through Protein–Polyelectrolyte Interaction: Kinetic Experiment and Molecular Dynamics Simulation. J. Phys. Chem. B. 2017, 121, 6785–6791.
- Reichert, C.; Borchard, G. Noncovalent PEGylation, An Innovative Subchapter in the Field of Protein Modification. J. Pharm. Sci. 2016, 105, 386–390.
- Ye, C.; Chi, H. A review of recent progress in drug and protein encapsulation: Approaches, applications and challenges. Mater. Sci. Eng. C 2018, 83, 233–246.
- Li, C.; Wu, G.; Ma, R.; Liu, Y.; Liu, Y.; Lv, J.; An, Y.; Shi, L. Nitrilotriacetic Acid (NTA) and Phenylboronic Acid (PBA) Functionalized Nanogels for Efficient Encapsulation and Controlled Release of Insulin. ACS Biomater. Sci. Eng. 2018, 4, 2007–2017.
- Chen, F.; Stenzel, M.H. Polyion Complex Micelles for Protein Delivery. Aust. J. Chem. 2018, 71, 768–780.
More
Information
Subjects:
Biochemical Research Methods
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
24 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No