 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vinod Labhasetwar | + 3533 word(s) | 3533 | 2022-02-21 07:39:15 | | | |
| 2 | Peter Tang | Meta information modification | 3533 | 2022-02-23 03:37:51 | | |
Video Upload Options
Free radicals are formed as a part of normal metabolic activities but are neutralized by the endogenous antioxidants present in cells/tissue, thus maintaining the redox balance. This redox balance is disrupted in certain neuropathophysiological conditions, causing oxidative stress, which is implicated in several progressive neurodegenerative diseases. Following neuronal injury, secondary injury progression is also caused by excessive production of free radicals. Highly reactive free radicals, mainly the reactive oxygen species (ROS) and reactive nitrogen species (RNS), damage the cell membrane, proteins, and DNA, which triggers a self-propagating inflammatory cascade of degenerative events. Dysfunctional mitochondria under oxidative stress conditions are considered a key mediator in progressive neurodegeneration. Exogenous delivery of antioxidants holds promise to alleviate oxidative stress to regain the redox balance.
1. Introduction
1.1. Endogenous and Exogenous Sources of Free Radicals
1.2. Free Radicals: A Double Edge Sword
2. Oxidative Stress and Neurodegenerative Diseases
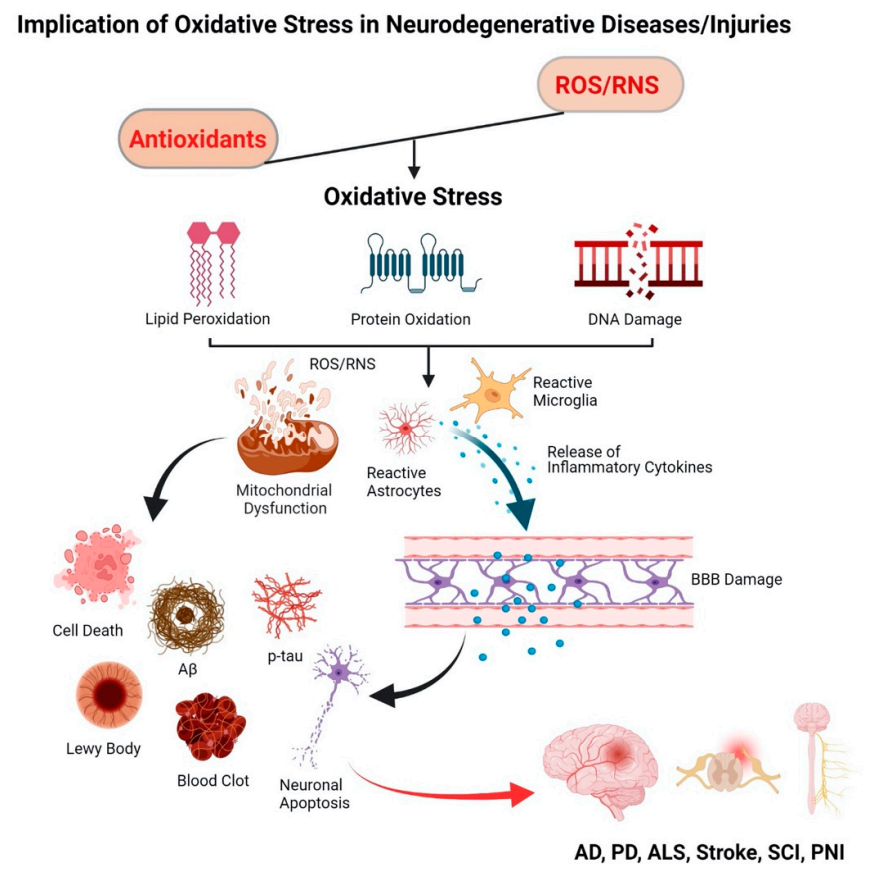
2.1. Progressive Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease (AD)
2.1.2. Parkinson’s Disease (PD)
2.1.3. Amyotrophic Lateral Sclerosis (ALS)
2.2. Injury-Induced Oxidative Stress
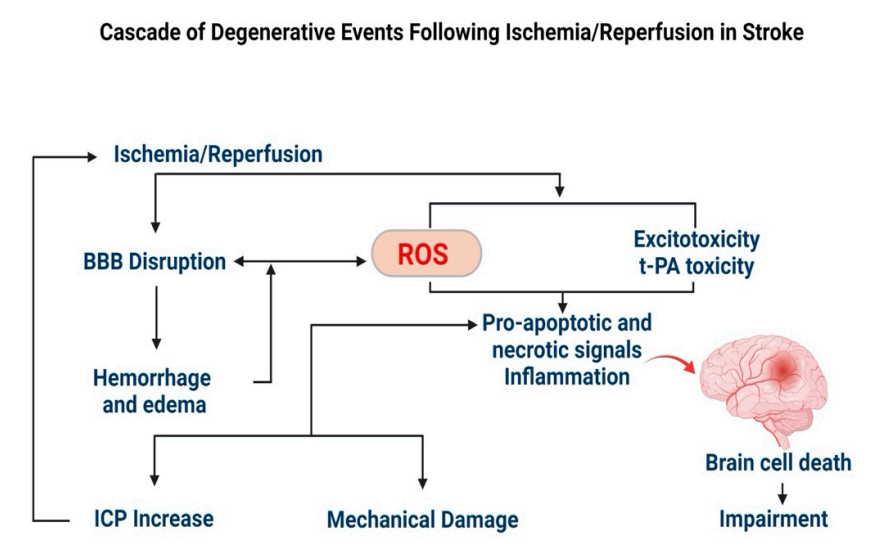
2.2.1. Stroke
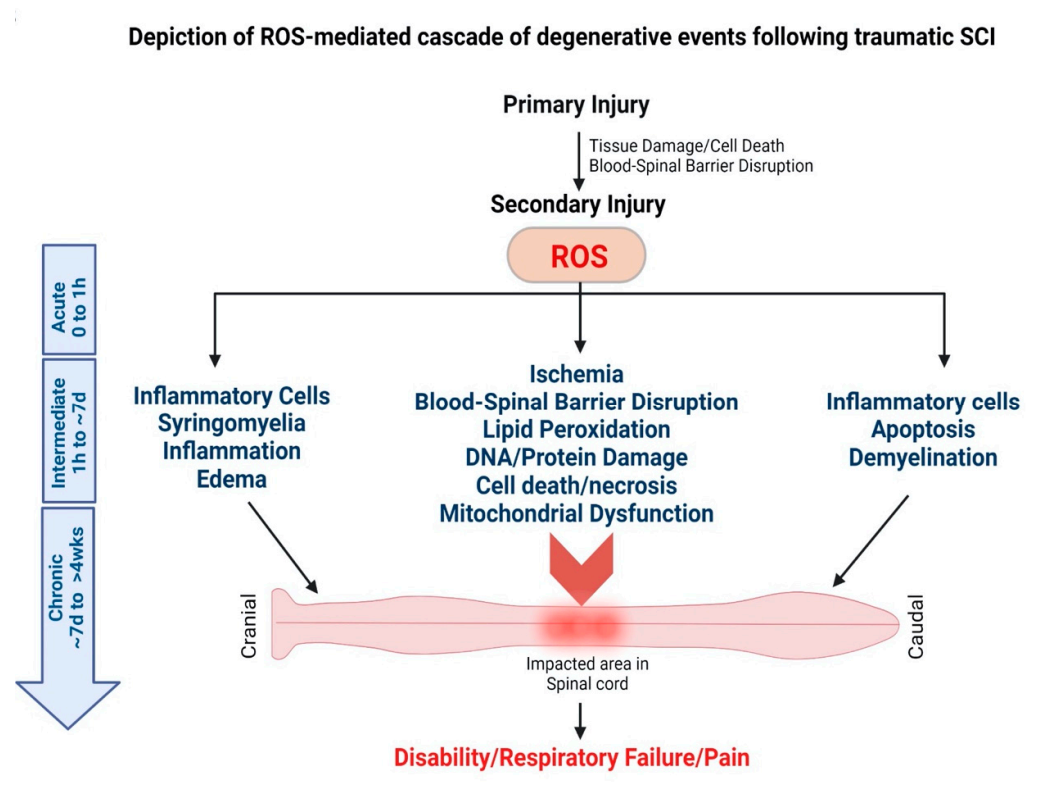
2.2.2. Spinal Cord Injury (SCI)
2.2.3. Peripheral Nerve Injury (PNI)
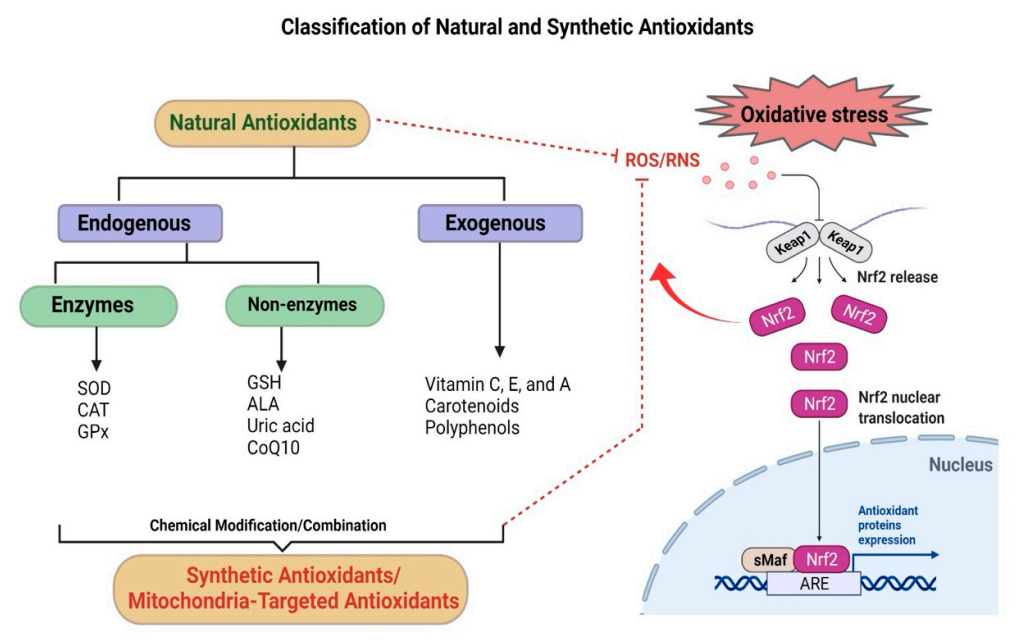
3. Antioxidants
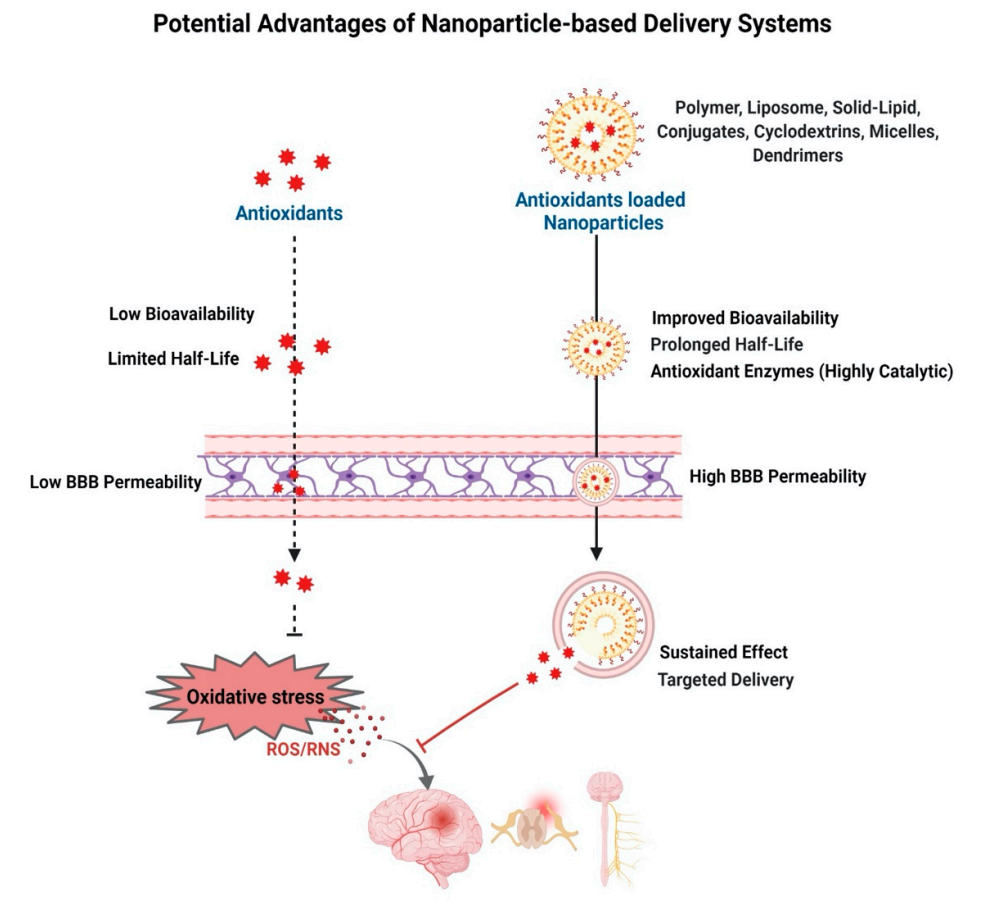
4. Antioxidant-Based Nanotherapy
References
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer Iv, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696.
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407.
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694.
- Man, A.W.C.; Li, H.; Xia, N. Impact of Lifestyles (Diet and Exercise) on Vascular Health: Oxidative Stress and Endothelial Function. Oxid. Med. Cell Longev. 2020, 2020, 1496462.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell Longev. 2020, 2020, 5021694.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Journey through the Diagnosis of Dementia—World Alzheimer Report 2021. Available online: https://www.alzint.org/resource/world-alzheimer-report-2021/ (accessed on 14 January 2022).
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533.
- von Arnim, C.A.; Herbolsheimer, F.; Nikolaus, T.; Peter, R.; Biesalski, H.K.; Ludolph, A.C.; Riepe, M.; Nagel, G.; Acti, F.E.U.S.G. Dietary antioxidants and dementia in a population-based case-control study among older people in South Germany. J. Alzheimers Dis. 2012, 31, 717–724.
- Urano, S.; Asai, Y.; Makabe, S.; Matsuo, M.; Izumiyama, N.; Ohtsubo, K.; Endo, T. Oxidative injury of synapse and alteration of antioxidative defense systems in rats, and its prevention by vitamin E. Eur. J. Biochem. 1997, 245, 64–70.
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901.
- Siraki, A.G.; O’Brien, P.J. Prooxidant activity of free radicals derived from phenol-containing neurotransmitters. Toxicology 2002, 177, 81–90.
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772.
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26.
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and brain diseases: The good, the bad, and the ugly. Oxid. Med. Cell Longev. 2013, 2013, 963520.
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput. Biol. 2011, 7, e1001115.
- Mailloux, R.J.; McBride, S.L.; Harper, M.E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602.
- Walker, C.L.; Pomatto, L.C.D.; Tripathi, D.N.; Davies, K.J.A. Redox Regulation of Homeostasis and Proteostasis in Peroxisomes. Physiol. Rev. 2018, 98, 89–115.
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981.
- Ballance, W.C.; Qin, E.C.; Chung, H.J.; Gillette, M.U.; Kong, H. Reactive oxygen species-responsive drug delivery systems for the treatment of neurodegenerative diseases. Biomaterials 2019, 217, 119292.
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51.
- Araujo, J.E.; Jorge, S.; Santos, H.M.; Chiechi, A.; Galstyan, A.; Lodeiro, C.; Diniz, M.; Kleinman, M.T.; Ljubimova, J.Y.; Capelo, J.L. Proteomic changes driven by urban pollution suggest particulate matter as a deregulator of energy metabolism, mitochondrial activity, and oxidative pathways in the rat brain. Sci. Total Environ. 2019, 687, 839–848.
- Calderon-Garciduenas, L.; Leray, E.; Heydarpour, P.; Torres-Jardon, R.; Reis, J. Air pollution, a rising environmental risk factor for cognition, neuroinflammation and neurodegeneration: The clinical impact on children and beyond. Rev. Neurol. 2016, 172, 69–80.
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148325.
- Mullenders, L.H.F. Solar UV damage to cellular DNA: From mechanisms to biological effects. Photochem. Photobiol. Sci. 2018, 17, 1842–1852.
- Akakin, D.; Tok, O.E.; Anil, D.; Akakin, A.; Sirvanci, S.; Sener, G.; Ercan, F. Electromagnetic Waves from Mobile Phones may Affect Rat Brain During Development. Turk. Neurosurg. 2021, 31, 412–421.
- Choi, S.; Krishnan, J.; Ruckmani, K. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86.
- Naha, N.; Gandhi, D.N.; Gautam, A.K.; Prakash, J.R. Nicotine and cigarette smoke modulate Nrf2-BDNF-dopaminergic signal and neurobehavioral disorders in adult rat cerebral cortex. Hum. Exp. Toxicol. 2018, 37, 540–556.
- Rodriguez-Martinez, E.; Nava-Ruiz, C.; Escamilla-Chimal, E.; Borgonio-Perez, G.; Rivas-Arancibia, S. The Effect of Chronic Ozone Exposure on the Activation of Endoplasmic Reticulum Stress and Apoptosis in Rat Hippocampus. Front. Aging Neurosci. 2016, 8, 245.
- Gargouri, B.; Bhatia, H.S.; Bouchard, M.; Fiebich, B.L.; Fetoui, H. Inflammatory and oxidative mechanisms potentiate bifenthrin-induced neurological alterations and anxiety-like behavior in adult rats. Toxicol. Lett. 2018, 294, 73–86.
- Lahouel, A.; Kebieche, M.; Lakroun, Z.; Rouabhi, R.; Fetoui, H.; Chtourou, Y.; Djamila, Z.; Soulimani, R. Neurobehavioral deficits and brain oxidative stress induced by chronic low dose exposure of persistent organic pollutants mixture in adult female rat. Environ. Sci. Pollut. Res. Int. 2016, 23, 19030–19040.
- Cruces-Sande, A.; Rodriguez-Perez, A.I.; Herbello-Hermelo, P.; Bermejo-Barrera, P.; Mendez-Alvarez, E.; Labandeira-Garcia, J.L.; Soto-Otero, R. Copper Increases Brain Oxidative Stress and Enhances the Ability of 6-Hydroxydopamine to Cause Dopaminergic Degeneration in a Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 2845–2854.
- Yauger, Y.J.; Bermudez, S.; Moritz, K.E.; Glaser, E.; Stoica, B.; Byrnes, K.R. Iron accentuated reactive oxygen species release by NADPH oxidase in activated microglia contributes to oxidative stress in vitro. J. Neuroinflamm. 2019, 16, 41.
- Langley, M.R.; Ghaisas, S.; Ay, M.; Luo, J.; Palanisamy, B.N.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Manganese exposure exacerbates progressive motor deficits and neurodegeneration in the MitoPark mouse model of Parkinson’s disease: Relevance to gene and environment interactions in metal neurotoxicity. Neurotoxicology 2018, 64, 240–255.
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci 2015, 143, 64–80.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049.
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. Biomed. Res. Int. 2014, 2014, 761264.
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295.
- Juan, C.A.; Perez de la Lastra, J.M.; Plou, F.J.; Perez-Lebena, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361.
- Singh, B.K.; Tripathi, M.; Pandey, P.K.; Kakkar, P. Alteration in mitochondrial thiol enhances calcium ion dependent membrane permeability transition and dysfunction in vitro: A cross-talk between mtThiol, Ca(2+), and ROS. Mol. Cell Biochem. 2011, 357, 373–385.
- Chiang, S.C.; Meagher, M.; Kassouf, N.; Hafezparast, M.; McKinnon, P.J.; Haywood, R.; El-Khamisy, S.F. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci. Adv. 2017, 3, e1602506.
- Eratne, D.; Loi, S.M.; Farrand, S.; Kelso, W.; Velakoulis, D.; Looi, J.C. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas. Psychiatry 2018, 26, 347–357.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147.
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231.
- Harman, D. Free radical theory of aging. Mutat. Res. 1992, 275, 257–266.
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8.
- Kim, S.H.; Vlkolinsky, R.; Cairns, N.; Lubec, G. Decreased levels of complex III core protein 1 and complex V beta chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell Mol. Life Sci. 2000, 57, 1810–1816.
- Kim, S.H.; Vlkolinsky, R.; Cairns, N.; Fountoulakis, M.; Lubec, G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer’s disease. Life Sci. 2001, 68, 2741–2750.
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446.
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 138–147.
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483.
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891.
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120.
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puolivali, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179.
- Mattson, M.P.; Fu, W.; Waeg, G.; Uchida, K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. Neuroreport 1997, 8, 2275–2281.
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588.
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S357–S367.
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44.
- Chen, L.; Na, R.; Gu, M.; Richardson, A.; Ran, Q. Lipid peroxidation up-regulates BACE1 expression in vivo: A possible early event of amyloidogenesis in Alzheimer’s disease. J. Neurochem. 2008, 107, 197–207.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12.
- Gomez-Benito, M.; Granado, N.; Garcia-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356.
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491.
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53.
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031.
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin Inhibits Activation of NADPH Oxidase/p38 MAPK Pathway and Enhances Expression of Antioxidant Protein in Parkinson Disease Models. Front. Mol. Neurosci. 2018, 11, 165.
- Park, H.R.; Yang, E.J. Oxidative Stress as a Therapeutic Target in Amyotrophic Lateral Sclerosis: Opportunities and Limitations. Diagnostics 2021, 11, 1546.
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509.
- Richardson, K.; Allen, S.P.; Mortiboys, H.; Grierson, A.J.; Wharton, S.B.; Ince, P.G.; Shaw, P.J.; Heath, P.R. The effect of SOD1 mutation on cellular bioenergetic profile and viability in response to oxidative stress and influence of mutation-type. PLoS ONE 2013, 8, e68256.
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345.
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10, 527.
- Obrador, E.; Salvador, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901.
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95.
- Bellezza, I.; Grottelli, S.; Costanzi, E.; Scarpelli, P.; Pigna, E.; Morozzi, G.; Mezzasoma, L.; Peirce, M.J.; Moresi, V.; Adamo, S.; et al. Peroxynitrite Activates the NLRP3 Inflammasome Cascade in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 2350–2361.
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxid. Med. Cell Longev. 2019, 2019, 1712323.
- Kazama, M.; Kato, Y.; Kakita, A.; Noguchi, N.; Urano, Y.; Masui, K.; Niida-Kawaguchi, M.; Yamamoto, T.; Watabe, K.; Kitagawa, K.; et al. Astrocytes release glutamate via cystine/glutamate antiporter upregulated in response to increased oxidative stress related to sporadic amyotrophic lateral sclerosis. Neuropathology 2020, 40, 587–598.
- Kolarcik, C.L.; Bowser, R. Retinoid signaling alterations in amyotrophic lateral sclerosis. Am. J. Neurodegener. Dis. 2012, 1, 130–145.
- Sun, M.S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.L.; Guo, Z.N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid. Med. Cell Longev. 2018, 2018, 3804979.
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317.
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting Oxidative Stress and Inflammation to Prevent Ischemia-Reperfusion Injury. Front. Mol. Neurosci. 2020, 13, 28.
- Al-Qazzaz, N.K.; Ali, S.H.; Ahmad, S.A.; Islam, S.; Mohamad, K. Cognitive impairment and memory dysfunction after a stroke diagnosis: A post-stroke memory assessment. Neuropsychiatr. Dis. Treat. 2014, 10, 1677–1691.
- Ahad, M.A.; Kumaran, K.R.; Ning, T.; Mansor, N.I.; Effendy, M.A.; Damodaran, T.; Lingam, K.; Wahab, H.A.; Nordin, N.; Liao, P.; et al. Insights into the neuropathology of cerebral ischemia and its mechanisms. Rev. Neurosci. 2020, 31, 521–538.
- Lorenzano, S.; Rost, N.S.; Khan, M.; Li, H.; Lima, F.O.; Maas, M.B.; Green, R.E.; Thankachan, T.K.; Dipietro, A.J.; Arai, K.; et al. Oxidative Stress Biomarkers of Brain Damage: Hyperacute Plasma F2-Isoprostane Predicts Infarct Growth in Stroke. Stroke 2018, 49, 630–637.
- Nakano, Y.; Yamashita, T.; Li, Q.; Sato, K.; Ohta, Y.; Morihara, R.; Hishikawa, N.; Abe, K. Time-dependent change of in vivo optical imaging of oxidative stress in a mouse stroke model. J. Neurosci. Res. 2017, 95, 2030–2039.
- Kishimoto, M.; Suenaga, J.; Takase, H.; Araki, K.; Yao, T.; Fujimura, T.; Murayama, K.; Okumura, K.; Ueno, R.; Shimizu, N.; et al. Oxidative stress-responsive apoptosis inducing protein (ORAIP) plays a critical role in cerebral ischemia/reperfusion injury. Sci. Rep. 2019, 9, 13512.
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089.
- Lou, Z.; Wang, A.P.; Duan, X.M.; Hu, G.H.; Song, G.L.; Zuo, M.L.; Yang, Z.B. Upregulation of NOX2 and NOX4 Mediated by TGF-beta Signaling Pathway Exacerbates Cerebral Ischemia/Reperfusion Oxidative Stress Injury. Cell Physiol. Biochem. 2018, 46, 2103–2113.
- Xu, N.; Meng, H.; Liu, T.; Feng, Y.; Qi, Y.; Wang, H. TRPC1 Deficiency Exacerbates Cerebral Ischemia/Reperfusion-Induced Neurological Injury by Potentiating Nox4-Derived Reactive Oxygen Species Generation. Cell Physiol. Biochem. 2018, 51, 1723–1738.
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835.
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507.
- Chen, S.; Shao, L.; Ma, L. Cerebral Edema Formation After Stroke: Emphasis on Blood-Brain Barrier and the Lymphatic Drainage System of the Brain. Front. Cell Neurosci. 2021, 15, 716825.
- van Den Hauwe, L.; Sundgren, P.C.; Flanders, A.E. Spinal Trauma and Spinal Cord Injury (SCI). In Diseases of the Brain, Head and Neck, Spine 2020–2023: Diagnostic Imaging; Hodler, J., Kubik-Huch, R.A., von Schulthess, G.K., Eds.; IDKD Springer Series: Cham, Switzerland, 2020.
- Eckert, M.J.; Martin, M.J. Trauma: Spinal Cord Injury. Surg. Clin. N. Am. 2017, 97, 1031–1045.
- Ahuja, C.S.; Nori, S.; Tetreault, L.; Wilson, J.; Kwon, B.; Harrop, J.; Choi, D.; Fehlings, M.G. Traumatic Spinal Cord Injury-Repair and Regeneration. Neurosurgery 2017, 80, S9–S22.
- Lin, J.; Xiong, Z.; Gu, J.; Sun, Z.; Jiang, S.; Fan, D.; Li, W. Sirtuins: Potential Therapeutic Targets for Defense against Oxidative Stress in Spinal Cord Injury. Oxid. Med. Cell Longev. 2021, 2021, 7207692.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Rabchevsky, A.G.; Michael, F.M.; Patel, S.P. Mitochondria focused neurotherapeutics for spinal cord injury. Exp. Neurol. 2020, 330, 113332.
- Popa, C.; Popa, F.; Grigorean, V.T.; Onose, G.; Sandu, A.M.; Popescu, M.; Burnei, G.; Strambu, V.; Sinescu, C. Vascular dysfunctions following spinal cord injury. J. Med. Life 2010, 3, 275–285.
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Qasim, M.; Zafar, S.; Aziz, N.; Razzaq, A.; Hussain, R.; de Aguilar, J.G.; et al. Current Status of Therapeutic Approaches against Peripheral Nerve Injuries: A Detailed Story from Injury to Recovery. Int. J. Biol. Sci. 2020, 16, 116–134.
- Walco, G.A.; Dworkin, R.H.; Krane, E.J.; LeBel, A.A.; Treede, R.D. Neuropathic pain in children: Special considerations. Mayo Clin. Proc. 2010, 85, S33–S41.
- Agbaje, J.O.; Van de Casteele, E.; Hiel, M.; Verbaanderd, C.; Lambrichts, I.; Politis, C. Neuropathy of Trigeminal Nerve Branches After Oral and Maxillofacial Treatment. J. Maxillofac. Oral Surg. 2016, 15, 321–327.
- Menorca, R.M.; Fussell, T.S.; Elfar, J.C. Nerve physiology: Mechanisms of injury and recovery. Hand Clin. 2013, 29, 317–330.
- Khezri, M.K.; Turkkan, A.; Koc, C.; Salman, B.; Levent, P.; Cakir, A.; Kafa, I.M.; Cansev, M.; Bekar, A. Anti-Apoptotic and Anti-Oxidant Effects of Systemic Uridine Treatment in an Experimental Model of Sciatic Nerve Injury. Turk. Neurosurg. 2021, 31, 373–378.
- Lattimore, M.R., Jr.; Varr, W.F., 3rd. Disposable soft lens ulcerative keratitis in an Army aviator: A case report. Aviat. Space Environ. Med. 1991, 62, 888–889.
- Costa, L.S.; Aidar, F.J.; Matos, D.G.; Oliveira, J.U.; Santos, J.L.D.; Almeida-Neto, P.F.; Souza, R.F.; Pereira, D.D.; Garrido, N.D.; Nunes-Silva, A.; et al. Effects of Resistance Training and Bowdichia virgilioides Hydroethanolic Extract on Oxidative Stress Markers in Rats Submitted to Peripheral Nerve Injury. Antioxidants 2020, 9, 941.
- Zafar, S.; Anwar, H.; Qasim, M.; Irfan, S.; Maqbool, J.; Sajid, F.; Naqvi, S.A.R.; Hussain, G. Calotropis procera (root) escalates functions rehabilitation and attenuates oxidative stress in a mouse model of peripheral nerve injury. Pak. J. Pharm. Sci. 2020, 33, 2801–2807.
- Tang, C.; Han, R.; Wu, J.; Fang, F. Effects of baicalin capsules combined with alpha-lipoic acid on nerve conduction velocity, oxidative stress and inflammatory injury in patients wi.ith diabetic peripheral neuropathy. Am. J. Transl. Res. 2021, 13, 2774–2783.
- Luca, M.; Luca, A.; Calandra, C. The Role of Oxidative Damage in the Pathogenesis and Progression of Alzheimer’s Disease and Vascular Dementia. Oxid. Med. Cell Longev. 2015, 2015, 504678.
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904.
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293.
- Oztop, D.; Altun, H.; Baskol, G.; Ozsoy, S. Oxidative stress in children with attention deficit hyperactivity disorder. Clin. Biochem 2012, 45, 745–748.
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68.
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67.
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today 2020, 25, 1270–1276.
- Geronzi, U.; Lotti, F.; Grosso, S. Oxidative stress in epilepsy. Expert Rev. Neurother. 2018, 18, 427–434.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695.
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184.
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014.
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206.
- Gerhke, S.A.; Shibli, J.A.; Salles, M.B. Potential of the use of an antioxidant compound to promote peripheral nerve regeneration after injury. Neural Regen. Res. 2015, 10, 1063–1064.
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell Longev. 2016, 2016, 2795090.
- Amato, A.; Terzo, S.; Mule, F. Natural Compounds as Beneficial Antioxidant Agents in Neurodegenerative Disorders: A Focus on Alzheimer’s Disease. Antioxidants 2019, 8, 608.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553.
- Vasconcelos, A.R.; Dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE Pathway Modulation by Dietary Energy Regulation in Neurological Disorders. Front. Pharmacol. 2019, 10, 33.
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution o.of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069.
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano 2010, 4, 6303–6317.
- De Matteis, V.; Rinaldi, R. Toxicity Assessment in the Nanoparticle Era. Adv. Exp. Med. Biol. 2018, 1048, 1–19.
- Karthikeyan, A.; Senthil, N.; Min, T. Nanocurcumin: A Promising Candidate for Therapeutic Applications. Front. Pharmacol. 2020, 11, 487.
- Fan, S.; Zheng, Y.; Liu, X.; Fang, W.; Chen, X.; Liao, W.; Jing, X.; Lei, M.; Tao, E.; Ma, Q.; et al. Curcumin-loaded PLGA-PEG nanoparticles conjugated with B6 peptide for potential use in Alzheimer’s disease. Drug Deliv. 2018, 25, 1091–1102.
- Fidelis, E.M.; Savall, A.S.P.; da Luz Abreu, E.; Carvalho, F.; Teixeira, F.E.G.; Haas, S.E.; Bazanella Sampaio, T.; Pinton, S. Curcumin-Loaded Nanocapsules Reverses the Depressant-Like Behavior and Oxidative Stress Induced by beta-Amyloid in Mice. Neuroscience 2019, 423, 122–130.
- Huo, X.; Zhang, Y.; Jin, X.; Li, Y.; Zhang, L. A novel synthesis of selenium nanoparticles encapsulated PLGA nanospheres with curcumin molecules for the inhibition of amyloid beta aggregation in Alzheimer’s disease. J. Photochem. Photobiol. B 2019, 190, 98–102.
- Ashafaq, M.; Intakhab Alam, M.; Khan, A.; Islam, F.; Khuwaja, G.; Hussain, S.; Ali, R.; Alshahrani, S.; Antar Makeen, H.; Alhazmi, H.A.; et al. Nanoparticles of resveratrol attenuates oxidative stress and inflammation after ischemic stroke in rats. Int. Immunopharmacol. 2021, 94, 107494.
- Lu, X.; Dong, J.; Zheng, D.; Li, X.; Ding, D.; Xu, H. Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia-reperfusion injury in rats. Nanomedicine 2020, 28, 102208.
- Kannan, S.; Dai, H.; Navath, R.S.; Balakrishnan, B.; Jyoti, A.; Janisse, J.; Romero, R.; Kannan, R.M. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 2012, 4, 130ra46.
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305.
- Yang, M.; Jin, L.; Wu, Z.; Xie, Y.; Zhang, P.; Wang, Q.; Yan, S.; Chen, B.; Liang, H.; Naman, C.B.; et al. PLGA-PEG Nanoparticles Facilitate In Vivo Anti-Alzheimer’s Effects of Fucoxanthin, a Marine Carotenoid Derived from Edible Brown Algae. J. Agric. Food Chem. 2021, 69, 9764–9777.
- Dhas, N.; Mehta, T. Cationic biopolymer functionalized nanoparticles encapsulating lutein to attenuate oxidative stress in effective treatment of Alzheimer’s disease: A non-invasive approach. Int. J. Pharm. 2020, 586, 119553.
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Inhibition of Al(III)-Induced Abeta42 Fibrillation and Reduction of Neurotoxicity by Epigallocatechin-3-Gallate Nanoparticles. J. Biomed. Nanotechnol. 2018, 14, 1147–1158.
- Liu, Z.; Li, X.; Wu, X.; Zhu, C. A dual-inhibitor system for the effective antifibrillation of Abeta40 peptides by biodegradable EGCG-Fe(iii)/PVP nanoparticles. J. Mater. Chem. B 2019, 7, 1292–1299.
- Liu, H.; Yu, L.; Dong, X.; Sun, Y. Synergistic effects of negatively charged hydrophobic nanoparticles and (-)-epigallocatechin-3-gallate on inhibiting amyloid beta-protein aggregation. J. Colloid Interface Sci. 2017, 491, 305–312.
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kuhne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75.
- Amin, F.U.; Shah, S.A.; Badshah, H.; Khan, M.; Kim, M.O. Anthocyanins encapsulated by nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Abeta1-42-induced oxidative stress. J. Nanobiotechnology 2017, 15, 12.
- Loureiro, J.A.; Andrade, S.; Duarte, A.; Neves, A.R.; Queiroz, J.F.; Nunes, C.; Sevin, E.; Fenart, L.; Gosselet, F.; Coelho, M.A.; et al. Resveratrol and Grape Extract-loaded Solid Lipid Nanoparticles for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 277.
- Amanzadeh Jajin, E.; Esmaeili, A.; Rahgozar, S.; Noorbakhshnia, M. Quercetin-Conjugated Superparamagnetic Iron Oxide Nanoparticles Protect AlCl3-Induced Neurotoxicity in a Rat Model of Alzheimer’s Disease via Antioxidant Genes, APP Gene, and miRNA-101. Front. Neurosci. 2020, 14, 598617.
- Halevas, E.; Mavroidi, B.; Nday, C.M.; Tang, J.; Smith, G.C.; Boukos, N.; Litsardakis, G.; Pelecanou, M.; Salifoglou, A. Modified magnetic core-shell mesoporous silica nano-formulations with encapsulated quercetin exhibit anti-amyloid and antioxidant activity. J. Inorg. Biochem. 2020, 213, 111271.
- Pinheiro, R.G.R.; Granja, A.; Loureiro, J.A.; Pereira, M.C.; Pinheiro, M.; Neves, A.R.; Reis, S. Quercetin lipid nanoparticles functionalized with transferrin for Alzheimer’s disease. Eur. J. Pharm. Sci. 2020, 148, 105314.
- Sun, D.; Li, N.; Zhang, W.; Zhao, Z.; Mou, Z.; Huang, D.; Liu, J.; Wang, W. Design of PLGA-functionalized quercetin nanoparticles for potential use in Alzheimer’s disease. Colloids Surf. B Biointerfaces 2016, 148, 116–129.
- Rifaai, R.A.; Mokhemer, S.A.; Saber, E.A.; El-Aleem, S.A.A.; El-Tahawy, N.F.G. Neuroprotective effect of quercetin nanoparticles: A possible prophylactic and therapeutic role in alzheimer’s disease. J. Chem. Neuroanat. 2020, 107, 101795.
- Moreno, L.; Puerta, E.; Suarez-Santiago, J.E.; Santos-Magalhaes, N.S.; Ramirez, M.J.; Irache, J.M. Effect of the oral administration of nanoencapsulated quercetin on a mouse model of Alzheimer’s disease. Int. J. Pharm. 2017, 517, 50–57.
- Ramires Junior, O.V.; Alves, B.D.S.; Barros, P.A.B.; Rodrigues, J.L.; Ferreira, S.P.; Monteiro, L.K.S.; Araujo, G.M.S.; Fernandes, S.S.; Vaz, G.R.; Dora, C.L.; et al. Nanoemulsion Improves the Neuroprotective Effects of Curcumin in an Experimental Model of Parkinson’s Disease. Neurotox. Res. 2021, 39, 787–799.
- Kundu, P.; Das, M.; Tripathy, K.; Sahoo, S.K. Delivery of Dual Drug Loaded Lipid Based Nanoparticles across the Blood-Brain Barrier Impart Enhanced Neuroprotection in a Rotenone Induced Mouse Model of Parkinson’s Disease. ACS Chem. Neurosci. 2016, 7, 1658–1670.
- Fernandes, E.J.; Poetini, M.R.; Barrientos, M.S.; Bortolotto, V.C.; Araujo, S.M.; Santos Musachio, E.A.; De Carvalho, A.S.; Leimann, F.V.; Goncalves, O.H.; Ramborger, B.P.; et al. Exposure to lutein-loaded nanoparticles attenuates Parkinson’s model-induced damage in Drosophila melanogaster: Restoration of dopaminergic and cholinergic system and oxidative stress indicators. Chem. Biol. Interact. 2021, 340, 109431.
- Palle, S.; Neerati, P. Improved neuroprotective effect of resveratrol nanoparticles as evinced by abrogation of rotenone-induced behavioral deficits and oxidative and mitochondrial dysfunctions in rat model of Parkinson’s disease. Naunyn. Schmiedebergs Arch. Pharmacol. 2018, 391, 445–453.
- Gaba, B.; Khan, T.; Haider, M.F.; Alam, T.; Baboota, S.; Parvez, S.; Ali, J. Vitamin E Loaded Naringenin Nanoemulsion via Intranasal Delivery for the Management of Oxidative Stress in a 6-OHDA Parkinson’s Disease Model. Biomed. Res. Int. 2019, 2019, 2382563.
- Trapani, A.; Guerra, L.; Corbo, F.; Castellani, S.; Sanna, E.; Capobianco, L.; Monteduro, A.G.; Manno, D.E.; Mandracchia, D.; Di Gioia, S.; et al. Cyto/Biocompatibility of Dopamine Combined with the Antioxidant Grape Seed-Derived Polyphenol Compounds in Solid Lipid Nanoparticles. Molecules 2021, 26, 916.
- Chen, L.; Watson, C.; Morsch, M.; Cole, N.J.; Chung, R.S.; Saunders, D.N.; Yerbury, J.J.; Vine, K.L. Improving the Delivery of SOD1 Antisense Oligonucleotides to Motor Neurons Using Calcium Phosphate-Lipid Nanoparticles. Front. Neurosci. 2017, 11, 476.
- Medina, D.X.; Chung, E.P.; Teague, C.D.; Bowser, R.; Sirianni, R.W. Intravenously Administered, Retinoid Activating Nanoparticles Increase Lifespan and Reduce Neurodegeneration in the SOD1(G93A) Mouse Model of ALS. Front. Bioeng. Biotechnol. 2020, 8, 224.
- Mauricio, M.D.; Guerra-Ojeda, S.; Marchio, P.; Valles, S.L.; Aldasoro, M.; Escribano-Lopez, I.; Herance, J.R.; Rocha, M.; Vila, J.M.; Victor, V.M. Nanoparticles in Medicine: A Focus on Vascular Oxidative Stress. Oxid. Med. Cell Longev. 2018, 2018, 6231482.
- Mei, T.; Kim, A.; Vong, L.B.; Marushima, A.; Puentes, S.; Matsumaru, Y.; Matsumura, A.; Nagasaki, Y. Encapsulation of tissue plasminogen activator in pH-sensitive self-assembled antioxidant nanoparticles for ischemic stroke treatment—Synergistic effect of thrombolysis and antioxidant. Biomaterials 2019, 215, 119209.
- Marques, M.S.; Cordeiro, M.F.; Marinho, M.A.G.; Vian, C.O.; Vaz, G.R.; Alves, B.S.; Jardim, R.D.; Hort, M.A.; Dora, C.L.; Horn, A.P. Curcumin-loaded nanoemulsion improves haemorrhagic stroke recovery in wistar rats. Brain Res. 2020, 1746, 147007.
- Chen, W.; Zhao, Z.; Zhao, S.; Zhang, L.; Song, Q. Resveratrol and Puerarin loaded polymeric nanoparticles to enhance the chemotherapeutic efficacy in spinal cord injury. Biomed. Microdevices 2020, 22, 69.
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763.
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi-Kashani, S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and Efficacy of Nanocurcumin as Add-On Therapy to Riluzole in Patients With Amyotrophic Lateral Sclerosis: A Pilot Randomized Clinical Trial. Neurotherapeutics 2018, 15, 430–438.
- NCT01001637; Efficacy and Safety of Curcumin Formulation in Alzheimer’s Disease. US National Library of Medicine: Bethesda, MD, USA, 2009.
- Cox, K.H.; Pipingas, A.; Scholey, A.B. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J. Psychopharmacol. 2015, 29, 642–651.
- Bao, Q.; Hu, P.; Xu, Y.; Cheng, T.; Wei, C.; Pan, L.; Shi, J. Simultaneous Blood-Brain Barrier Crossing and Protection for Stroke Treatment Based on Edaravone-Loaded Ceria Nanoparticles. ACS Nano 2018, 12, 6794–6805.
- Jin, Q.; Cai, Y.; Li, S.; Liu, H.; Zhou, X.; Lu, C.; Gao, X.; Qian, J.; Zhang, J.; Ju, S.; et al. Edaravone-Encapsulated Agonistic Micelles Rescue Ischemic Brain Tissue by Tuning Blood-Brain Barrier Permeability. Theranostics 2017, 7, 884–898.
- Wang, G.Y.; Rayner, S.L.; Chung, R.; Shi, B.Y.; Liang, X.J. Advances in nanotechnology-based strategies for the treatments of amyotrophic lateral sclerosis. Mater. Today Bio 2020, 6, 100055.
- Dang, L.; Dong, X.; Yang, J. Influence of Nanoparticle-Loaded Edaravone on Postoperative Effects in Patients with Cerebral Hemorrhage. J. Nanosci. Nanotechnol. 2021, 21, 1202–1211.

