Free radicals are formed as a part of normal metabolic activities but are neutralized by the endogenous antioxidants present in cells/tissue, thus maintaining the redox balance. This redox balance is disrupted in certain neuropathophysiological conditions, causing oxidative stress, which is implicated in several progressive neurodegenerative diseases. Following neuronal injury, secondary injury progression is also caused by excessive production of free radicals. Highly reactive free radicals, mainly the reactive oxygen species (ROS) and reactive nitrogen species (RNS), damage the cell membrane, proteins, and DNA, which triggers a self-propagating inflammatory cascade of degenerative events. Dysfunctional mitochondria under oxidative stress conditions are considered a key mediator in progressive neurodegeneration. Exogenous delivery of antioxidants holds promise to alleviate oxidative stress to regain the redox balance.
- neurodegeneration
- reactive oxygen species
- inflammation
- polymers
- CNS
- antioxidant enzymes
1. Introduction
1.1. Endogenous and Exogenous Sources of Free Radicals
1.2. Free Radicals: A Double Edge Sword
2. Oxidative Stress and Neurodegenerative Diseases
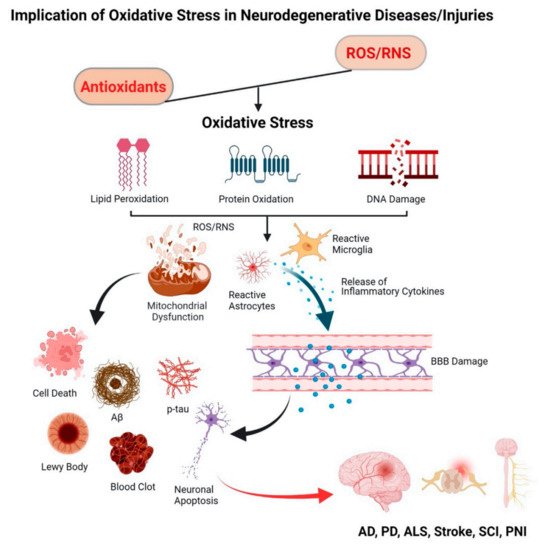
2.1. Progressive Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease (AD)
2.1.2. Parkinson’s Disease (PD)
2.1.3. Amyotrophic Lateral Sclerosis (ALS)
2.2. Injury-Induced Oxidative Stress
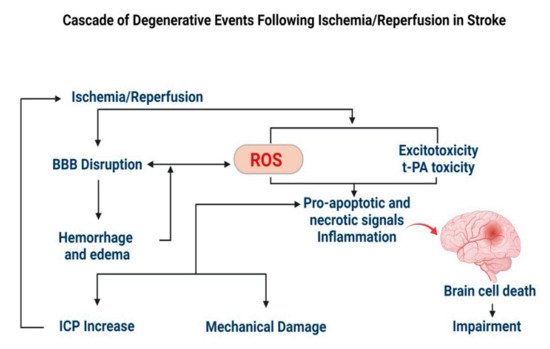
2.2.1. Stroke
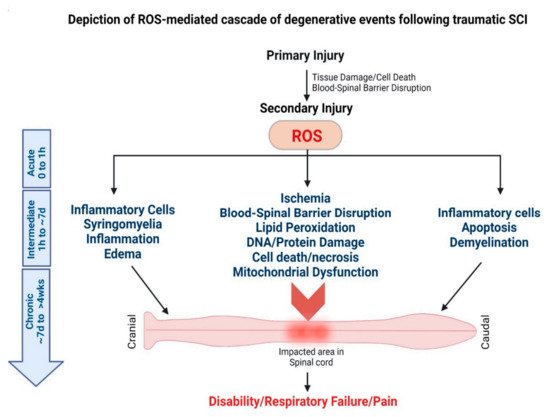
2.2.2. Spinal Cord Injury (SCI)
2.2.3. Peripheral Nerve Injury (PNI)
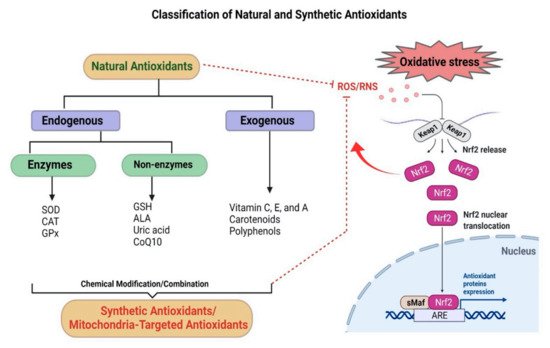
3. Antioxidants
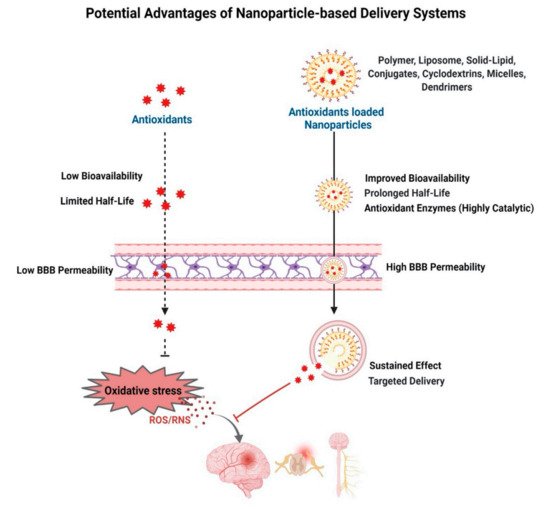
4. Antioxidant-Based Nanotherapy
This entry is adapted from the peer-reviewed paper 10.3390/antiox11020408