 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eduardo Pena | + 2841 word(s) | 2841 | 2020-09-08 07:52:40 | | | |
| 2 | Vicky Zhou | -19 word(s) | 2822 | 2020-10-27 09:42:39 | | |
Video Upload Options
Under hypobaric hypoxia contidition, the principal impact on humans is due to the low atmospheric pressure and the subsequent proportional decrease in partial oxygen pressure (PO2) in the inspired air, generating a reduction in the bioavailability of oxygen in organs, tissues and cells at high altitude.
1. Introduction
Many people are exposed to high altitudes, where more than 140 million of them live permanently at an elevation > 2500 m above sea level, and approximately 40 million individuals are exposed to high altitudes for hours or days[1]. It is important to highlight that high altitude presents an extreme environment (i.e., extreme temperatures, low air humidity and high ultraviolet radiation level), which is challenging for the human body, making it very difficult to adapt to this condition [2][3]. Some ethnic groups, such as the Tibetan people, have undergone adaptive changes in their metabolism[4].
The classification according to the time of exposure to hypobaric hypoxia is very important, since there are three well-determined conditions: chronic hypobaric exposure that represents people who live permanently at high altitude (Andes and Himalayas)[5] and acute exposure that represents people who are exposed to high altitude for hours or days (tourists and climbers). However, in recent years, a new condition associated with high altitude has been reported in South America as long-term chronic intermittent hypobaric hypoxia; this type of exposure refers to people commuting to work at high altitudes for several days and returning to sea level to rest for the same number of days for years. This condition is termed the “Chilean miner model” of exposure to chronic intermittent hypobaric hypoxia[6].
One of the principal pathologies developed under hypobaric hypoxia, both chronic and intermittent, is high-altitude pulmonary hypertension (HAPH)[7][8]. HAPH is produced initially by hypoxic pulmonary vasoconstriction (HPV), which begins as a compensatory mechanism to distribute the blood to more ventilated areas of the lung[9], mediated through calcium release that acts on Rho-associated protein kinase and the actin-myosin contractile apparatus[10]. Additionally, the activation of molecular pathways and morphological changes, such as uncoupled endothelial nitric oxide synthase (eNOS), pro-inflammatory cytokines such as interleukin 1 and 6 (IL-1 and IL-6), and oxidative stress, among others, lead to pulmonary arterial remodeling and ultimately definitive pulmonary hypertension (PH)[11][12]. Thus, the principal consequence of HAPH is pressure overload (PO)-induced right ventricular hypertrophy (RVH) in the long term[13][14].
Cardiac hypertrophy is defined as an enlargement of the heart wall with an increase in the volume of cardiomyocytes; there are two forms of cardiac hypertrophy, namely, physiological, as occurs in response to exercise, and pathological, as occurs in response to abnormal stress, such as hypertension, pressure overload, endocrine disorders, myocardial infarction, and contractile dysfunction from inherited mutations in sarcomeric or cytoskeletal proteins[15][16]. Initially, this acclimatization response allows normal wall stress function, preserving right ventricle (RV) function, but in the long term, this acclimatization hypertrophy mechanism is overrun; thus, contractile dysfunction and RV dilatation occur, with a subsequent increase in wall stress that stimulates further hypertrophy, leading to a vicious cycle in the impairment of RV performance, which derives in RV failure and eventually leads to death[17][18][19].
Despite the large amount of information that explains RVH by PH-induced PO, we cannot ignore that hypoxia triggers the activation of several molecular pathways that could contribute to both the hypertrophic effect and the dilation process in RV. In fact, studies under hypoxic conditions have determined the important role of oxidative stress[20][21], kinase activation[22][23] and inflammatory processes[24] as possible contributors to RVH development and to the transition to right heart failure (RHF)[25]. However, it is important to highlight that the control mechanisms of RVH and heart failure (HF) in hypobaric hypoxia are still not well understood, and cardiac-directed therapies are rather scarce[23]. Therefore, the aim is to describe and update the knowledge on the molecular pathways involved in RVH and RHF under hypobaric hypoxia, including oxidative stress, protein kinase activation and inflammatory processes, as well as the current therapeutic approaches for these conditions.
2. Oxidative stress, inflammation and kinase activity
The reactive oxygen species (ROS) molecules play an important role in several cardiac physiological processes, such as the regulation of heart development and cardiomyocyte maturation, cardiac calcium handling[26], as well as excitation and contraction coupling[27][28]. However, when the level of ROS increases due to environmental or chemical stimuli and is not compensated by the endogenous antioxidant system, such as superoxide dismutases (SODs), catalase (CAT), glutathione peroxidase/reductase (GSH-PX/RX) and peroxiredoxin/thioredoxin[28], oxidative stress is triggered. Moreover, studies indicate that oxidative stress and inflammation are closely associated with the progression of myocardial hypertrophy and myocardial infarction[29][30][31]. Studies suggest that oxidative stress can activate nuclear factor-kappa B (NF-kB) in cardiomyocytes, a transcription factor implicated in the regulation of the inflammatory response, which promotes cardiac remodeling and failure[32][33].
Many studies in both humans and animals show an increase in ROS in plasma, lipids and tissues after exposure to acute or chronic hypobaric hypoxia (3600–7620 m, 13–8% O2)[2][21][34][35][36]. Further support is provided by studies of murine hearts under acute hypobaric hypoxia (10 h, 4570 m, 12% O2), showing an upregulation in the transcription levels of pro-oxidant molecules[37]. In other words, oxidative stress under any condition of hypoxia triggers redox signaling pathways in the heart, which in some cases is related to the transcription factor induced by hypoxia, termed hypoxia-inducible factor (HIF)-1α, which will be described in detail later.
Cardiac hypertrophy has also been a focus as a consequence of the role of the superfamily of mitogen-activated protein kinases (MAPKs) as important mediators of hypertrophic stimuli in the heart[22][38]. It is important to highligh that JNK, p38 and ERK are considered redox-sensitive kinases, and their activity is usually reported to regulate inflammation and apoptosis of cardiomyocytes[39][40].
Although studies have shown an important role of p38 and JNK in cardiac hypertrophy[41][42], very few studies have been conducted under hypobaric hypoxia conditions. Some studies regarding the activity of JNK and p38 in rats showed an inactive state of these proteins in the heart, specifically in RVH, under both intermittent hypobaric hypoxia (8 h/d, 5 days, 7000 m) and acute hypobaric hypoxia (9 h, 8000 m) exposure[43][44]. Conversely, p38 presents four isoforms, α, β, γ (also known as Erk6 or SAPK3), and δ (also known as SAPK4), and their expression depends on the tissue and the cardiac process, since only p38α and p38β2 are related to cardiac ventricular hypertrophy[45]. In addition, studies have demonstrated that under another hypoxic condition (chronic normobaric hypoxia), p38, specifically the α subunit, could have a role in heart failure, as will be discussed later.
Likewise, studies in mice with RVH induced by hypobaric hypoxia (5380 m, 10% O2) showed a relation with MAP kinase kinase kinase-2 (MEKK2) and the ERK5 pathway, where MEKK2 can coordinately activate signals through MEK5/ERK5 protein kinases, leading to an increase in inflammatory molecules such as interleukin 1β (IL-1β), stromal cell-derived factor 1 (SDF-1), monocyte chemoattractant protein-1 (MCP-1) and C-X-C chemokine receptor type 4 (CXCR-4) triggering the hypertrophic process[23]. In addition, studies have shown an increase in H2O2-induced mirR-143-3p expression, which increases ERK5 activity in myocardial hypertrophy[46]. However, this is not the only kinase involved in this condition. Hypoxia exposure produces an alteration of protein kinase C (PKC), considered a key regulator in PO-induced cardiac hypertrophy, and its activation can be modulated by ROS derived from mitochondrial complex III[47]. Additionally, it is important to highlight that, similar to p38, PKC has different isoforms (α, β, ε, δ, γ, η, θ and ζ), and their expression depends on factors such as hypoxic stimuli[48], zinc administration[49] and ROS[50].
Consequently, studies in rats with RVH induced by hypobaric hypoxia (380 mmHg, 5500 m) have shown upregulation and activation of PKC-α only in RV[48]. Interestingly, later studies showed that PKC-α produces an increase in galectin-3 expression, which subsequently promotes cardiac fibrosis and HF[51]. Therefore, the results are interesting to determine in future studies the activation and contribution of this PKC-α-galectin-3 pathway in RVH under hypobaric hypoxia conditions. On the other hand, there are some crucial events that characterize this transition from compensatory cardiac hypertrophy to decompensated hypertrophy and HF[52]. The most relevant events in this transition are cardiac fibrosis (through the differentiation of cardiac fibroblasts to myofibroblasts), apoptosis and inflammation[24][25], all of them being associated with oxidative stress.
2.1. Fibrosis
Supporting the role of fibrosis, a recent study in rats with RVH induced by hypobaric hypoxia (4500 m) exposure showed a fibrotic process in the RV[53]. A study in mice under other types of hypoxia (chronic normobaric hypoxia, 10% O2) showed that animals with PO-induced RVH and HF exhibited increased p38α expression, which was related to several effects, such as increased collagen and smooth muscle actin α (α-SMA) content leading to fibrosis[54]. Additionally, it is important to highlight that p38α is necessary for the differentiation of cardiac fibroblasts to myofibroblasts through the nuclear translocation of myocardin-related transcription factor A (MRTF-A)[54]. However, there are other molecules induced by hypoxia, such as IL-6, endothelin-1 (ET-1) and transforming growth factor α and β (TGF-α and TGF-β), that are related to the transdifferentiation of fibroblasts to myofibroblasts[55], highlighting the key role of IL-6[56], as will be discussed later.
Myofibroblasts are not part of normal cardiac tissue and appear only following cardiac injury[57]. This type of cell is considered a contractile cell due to the generation of bridges between the internal microfilament and the extracellular fibronectin domains of the myofibroblast through a specialized adhesion complex termed the fibronexus, functioning as a contractile mechanism that produces a force on the surrounding extracellular matrix[57]. Myofibroblasts can also produce and secrete several cytokines, chemokines and growth factors (IL-1α and β, IL-6, IL-10, TNF-α, IL-8, MCP-1, TGF-β, among others) by themselves, which are key in cellular events that lead to inflammatory and fibrotic responses during cardiac stress [58]. It is important to highlight that like fibroblasts, myofibroblasts are not excitable cells; therefore, the increased number of these cells decreases myocyte-to-myocyte coupling through gap junctions[57], which produces a barrier between cardiomyocytes contributing to the development of HF; however, more studies are needed.
Regarding collagen, studies focused on cardiac fibroblasts of RV have shown activation of PKCβ2 and PKCδ, producing an increase in collagen content under hypoxic (10% O2) conditions[59]. On the other hand, it has been demonstrated that hypoxia-modulated ORAI1 channels (a nonvoltage-gated channel) that regulate extracellular Ca2+ entry through the stromal interacting molecule-1 (STIM1) protein might contribute to RVH and RHF. In addition, these proteins are also expressed in cardiac fibroblasts contributing to cardiac fibrosis[19].
2.2. Inflammation
As mentioned above, another important factor is inflammation associated with elevated expression of cytokines, chemokines and transcription factors, such as IL-1β, IL-6, tumor necrosis factor α (TNF-α), MCP-1 and NF-kB, which might lead to right ventricle dysfunction and failure[24] and might also be present in RVH. MCP-1 plays an important role in monocyte recruitment-induced inflammation in cardiac tissue, since studies show that monocyte recruitment is tightly regulated by the interaction between MCP-1 and its receptor CCR2 in myocardial damage[60][61].
Several studies have described that in different PH models, the inflammatory molecule IL-6 is increased both in plasma and RV tissue, which could be involved in cardiac function and morphology[33]. This is supported by a study by Nehra et al.[14] in rats with RVH induced by hypobaric hypoxia (7620 m) showing an increase in circulating levels of TNF-α and IL-6, and these molecules were associated with a decompensated transition to RHF. Moreover, a recent study in mice with transverse aortic constriction shows new insight into hypoxia-induced mitogenic factor (HIMF), a cytokine-like protein that could induce cardiac hypertrophy, fibrosis and myofibroblast differentiation through IL-6[62], which results in an interesting pathway to be analyzed in the transition to HF under this particular condition. However, a study in 15 healthy humans (age: 39 ± 10.2; 4 women, 11 men; BIM 23.6 ± 2) of European ancestry exposed for 72 h at 3830 m (acute hypobaric hypoxia) determined no changes in the plasma levels of IL-1β, IL-6 and NF-kB. However, the ROS production rate (O2·− and ·OH) and markers of ROS damage, such as lipid peroxidation, 8-isoPGF2α, 8-hydroxy-2′-deoxyguanosine (products of DNA oxidation) and protein oxidative carbonylation, were significantly increased over time[21].
2.3. Apoptosis
Regarding apoptotic events, studies have shown that hypobaric hypoxia exposure produces an increase in cardiac apoptosis in both rat and mouse models[14][63]. In addition, studies have shown that oxidative stress may further accelerate the development of RHF through the induction of more cardiac apoptosis[64]. In addition, cytokines, such as TNF-α, are also involved in the apoptotic process in cardiomyocytes[65]. It is important to highlight that this molecule is increased by ROS[66]; therefore, we can hypothesize that hypoxia-induced ROS could trigger cardiac apoptosis through TNF-α. This is supported by recent studies in mice and rats that demonstrated that the administration of ROS scavenger molecules, such as nitronyl nitroxide radicals, curcumin and nanocurcumin, under hypobaric hypoxia conditions, prevents oxidative stress and attenuates cardiac apoptosis[14][63].
2.4. HIF-1α
It is important to note that several cytokines and inflammatory processes, such as inflammatory cell migration (monocytes and macrophages) and monocyte differentiation to macrophages, are modulated by HIF-1α under hypoxic conditions [67][68]. Moreover, monocyte maturation to macrophages is accompanied by the production of cytokines and growth factors[61], since macrophages contribute to the release of different cytokines, such as TNF-α, IL-1β, IL-6, IL-12 and IL-23[24]. In addition, the cytokine TNF-α produced by macrophages stabilizes and increases the activity of HIF-1α, generating a positive feedback that leads to further differentiation of monocytes to macrophages[68]. It is important to mention that NF-kB is a key mediator in TNF-α-induced HIF-1α stabilization and activation[68]; hence, the relation between hypoxia-induced factors, such as HIF-1α, and inflammatory pathways in macrophages are potentiated. However, the role of HIF-1 in cardiac hypertrophy and HF remains to be determined[67]. A proposed schematic in cardiac myocytes summarizing all these molecular pathways is depicted in Figure 1.
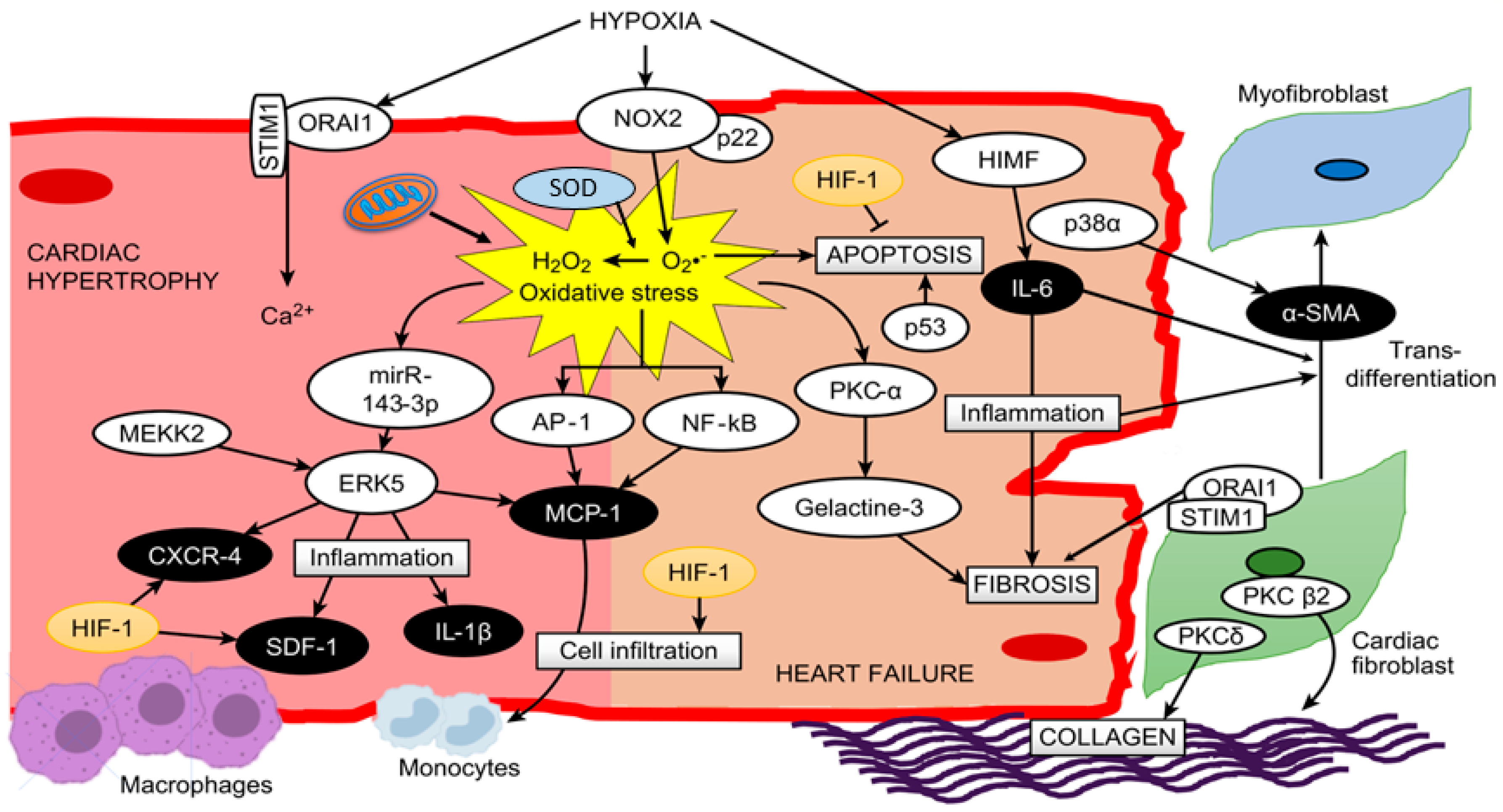
Figure 1. A proposed scheme summarizing all described molecular pathways and cells involved in cardiac hypertrophy and heart failure under hypoxia.
3. Treatment Approaches
3.1. Phytotherapy
Based on the important role of oxidative stress, fibrosis and inflammatory processes, current studies have focused on new therapies to limit ROS production and enhance ROS detoxification as means of ameliorating cardiac disease outcomes[28]. This can be supported by studies in rats exposed to acute hypobaric hypoxia (8000 m), which showed that the administration of an ancestral medicine of Tibet (Rhodiola crenulata) for preventing high-altitude illness decreases arginase 1 activity, oxidative stress and apoptosis and increases eNOS activity in heart tissue[44].
Studies by Nehra et al.[14] have demonstrated that nanocurcumin protects the heart against hypobaric hypoxia effects even better than curcumin. Additionally, the combination of preconditioning physical training plus oral administration of antioxidant nanocurcumin formulation decreases the RVH, circulatory markers of cardiac damage and oxidative stress in rats exposed to hypobaric hypoxia (7620 m, 8% O2) even more than each treatment alone[69]. Finally, the administration of Hippophae rhamnoides L. extract reduced oxidative stress (malondialdehyde level), fibrosis and NF-kB expression in the hearts of rats exposed to hypobaric hypoxia[70].
3.2. Pharmacological and Gene Therapy
Studies in transgenic mice with hypobaric hypoxia-induced RVH showed that target disruption of MEKK2, through insertion of the neomycin resistance gene in the coding sequence, attenuates ERK5 abundance and activation, which is consistent with an attenuation of RVH[23]. It is important to highlight that in this study, a reduction in inflammatory molecules related to the cardiac hypertrophy process, such as IL-1β, MCP-1, SDF-1 and CXCR-4, was also observed[23]. Another interesting approach to improve cardiac outcomes is the use of hemoglobin-based oxygen carriers to deliver carbon monoxide in mice exposed to intermittent hypobaric hypoxia (8 h/day, 7 d; 5500 m), which showed improved cardiac function, reduced myocardial apoptosis, and attenuated mitochondrial oxidative stress under treatment, highlighting an anti-inflammatory effect through a decrease in IL-6 and TNF-α levels in plasma[71].
A study in rats exposed to hypobaric hypoxia (5500 m, 380 mm Hg) with administration of salubrinal, a specific inhibitor of eIF2α phosphatase enzyme, showed that this molecule ameliorates RVH and cardiac fibrosis and attenuates oxidative stress and ET-1 levels in serum[72]. All these treatments mentioned would contribute to further support of the pivotal role of ROS, TNF-α and IL-6 molecules in the transition from compensatory cardiac hypertrophy to decompensated hypertrophy and HF under hypobaric hypoxia conditions.
On the other hand, a recent study in rats exposed to hypobaric hypoxia (10% O2) administered soluble macromolecules termed drag-reducing polymers and determined that these drugs attenuate right ventricular dysfunction and hypertrophy[73]. A recent study showed that the administration of atorvastatin increases PKCε, increasing its cardioprotective effects through the inhibition of miR-31 under intermittent hypoxia (5% O2) conditions[74]. This therapy opens a promising avenue to attempt the search for molecules to block deleterious pathways or induce protective pathways. Finally, these pharmacological treatments, either alone or combined, under hypoxic conditions seem to be very challenging. A proposed scheme of the phytotherapy and pharmacological approaches is depicted in Figure 2.
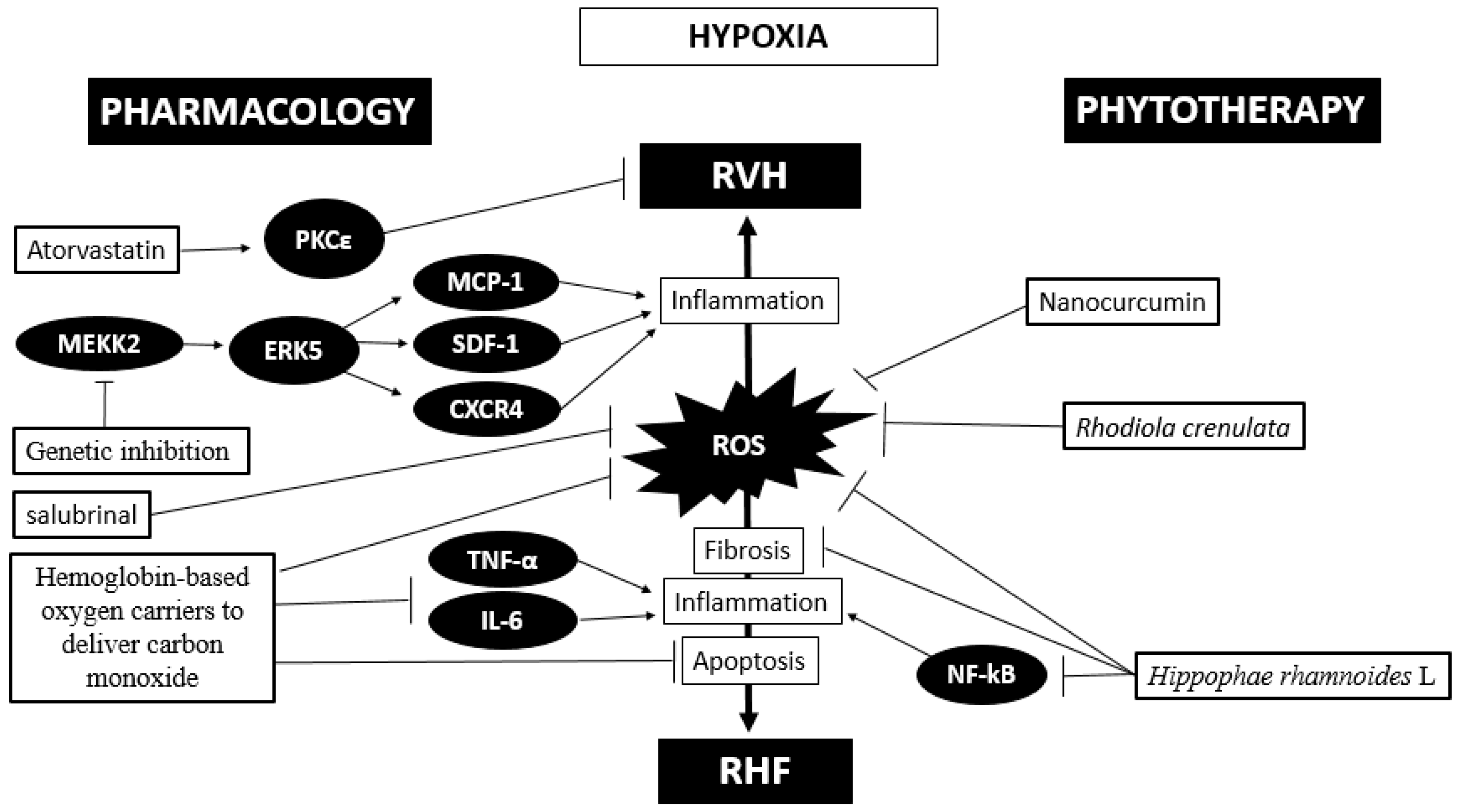
In summary, oxidative stress plays a pivotal role in promoting the activation of some kinases in inducing an inflammatory status in hypobaric hypoxia-induced RVH and HF. Pharmacological therapy, phytotherapy and gene therapy have been attempted with promising results that need to be validated.
References
- Aibek E. Mirrakhimov; Kingman P. Strohl; High-altitude Pulmonary Hypertension: an Update on Disease Pathogenesis and Management. The Open Cardiovascular Medicine Journal 2016, 10, 19-27, 10.2174/1874192401610010019.
- Akanksha Agrawal; Richa Rathor; Geetha Suryakumar; Oxidative protein modification alters proteostasis under acute hypobaric hypoxia in skeletal muscles: a comprehensive in vivo study. Cell Stress and Chaperones 2017, 22, 429-443, 10.1007/s12192-017-0795-8.
- Grzegorz Bilo; Sergio Caravita; Camilla Torlasco; Gianfranco Parati; Blood pressure at high altitude: physiology and clinical implications.. Kardiologia Polska 2019, 77, 596-603, 10.33963/KP.14832.
- Cynthia M. Beall; Daniel Laskowski; Serpil C. Erzurum; Nitric oxide in adaptation to altitude. Free Radical Biology and Medicine 2012, 52, 1123-1134, 10.1016/j.freeradbiomed.2011.12.028.
- Moore, LG.; Human genetic adaptation to high altitude. High Alt Med Biol. 2001, 2, 257-279, 10.1089/152702901750265341.
- Jean-Paul Richalet; Manuel Vargas Donoso; Daniel Jimenez; Ana-María Antezana; Cristián Hudson; Guillermo Cortes; Jorge Osorio; Angelica Leon; Chilean Miners Commuting from Sea Level to 4500 m: A Prospective Study. High Altitude Medicine & Biology 2002, 3, 159-166, 10.1089/15270290260131894.
- Fabiola León-Velarde; Marco Maggiorini; John T. Reeves; A. A. Aldashev; Ingrid Asmus; Luciano Bernardi; Ri-Li Ge; Peter Hackett; Toshio Kobayashi; Lorna G. Moore; et al.Dante PenalozaJean-Paul RichaletRobert C. RoachTianyi WuEnrique VargasGustavo Zubieta-CastilloGustavo Zubieta-Calleja Consensus Statement on Chronic and Subacute High Altitude Diseases. High Altitude Medicine & Biology 2005, 6, 147-157, 10.1089/ham.2005.6.147.
- M.M. Hoeper; Marc Humbert; Rogerio Souza; Majdy Idrees; Steven M Kawut; Karen Sliwa; Zhi-Cheng Jing; J Simon R Gibbs; A global view of pulmonary hypertension. The Lancet Respiratory Medicine 2016, 4, 306-322, 10.1016/s2213-2600(15)00543-3.
- U. S. V. Euler; G. Liljestrand; Observations on the Pulmonary Arterial Blood Pressure in the Cat. Acta Physiologica Scandinavica 1946, 12, 301-320, 10.1111/j.1748-1716.1946.tb00389.x.
- Rohit Moudgil, Evangelos D Michelakis, Stephen L Archer; Hypoxic pulmonary vasoconstriction. J Appl Physiol 2005, 1, 390-403, 1152/japplphysiol.00733.2004.
- N F Voelkel; R M Tuder; James P. Bridges; W P Arend; Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline.. American Journal of Respiratory Cell and Molecular Biology 1994, 11, 664-675, 10.1165/ajrcmb.11.6.7946395.
- Patricia Siques; Julio Brito; Eduardo Pena; Reactive Oxygen Species and Pulmonary Vasculature During Hypobaric Hypoxia. Frontiers in Physiology 2018, 9, 865, 10.3389/fphys.2018.00865.
- Dante Penaloza; Javier Arias-Stella; Marcelo V. Elizari; Rafael S. Acunzo; Marcela Ferreiro; The Heart and Pulmonary Circulation at High Altitudes. Circulation 2007, 115, 1132-1146, 10.1161/circulationaha.106.624544.
- Sarita Nehra; Varun Bhardwaj; Santosh Kar; Deepika Saraswat; Chronic Hypobaric Hypoxia Induces Right Ventricular Hypertrophy and Apoptosis in Rats: Therapeutic Potential of Nanocurcumin in Improving Adaptation. High Altitude Medicine & Biology 2016, 17, 342-352, 10.1089/ham.2016.0032.
- Eric N. Olson; Michael D. Schneider; Sizing up the heart: development redux in disease. Genes & Development 2003, 17, 1937-1956, 10.1101/gad.1110103.
- Sheng-Huang Chang; Chung-Jung Liu; Chia-Hua Kuo; Hong Chen; Wen-Yuan Lin; Kun-Yu Teng; Chang-Hai Tsai; Fuu-Jen Tsai; Chih-Yang Huang; Bor-Show Tzang; et al.Wei-Wen KuoSheng-Wei Chang Garlic Oil Alleviates MAPKs- and IL-6-mediated Diabetes-related Cardiac Hypertrophy in STZ-induced DM Rats. Evidence-Based Complementary and Alternative Medicine 2010, 2011, 1-11, 10.1093/ecam/neq075.
- Norbert F. Voelkel; Robert A. Quaife; Leslie A. Leinwand; Robyn J. Barst; Michael D. McGoon; Daniel R. Meldrum; Jocelyn Dupuis; Carlin S. Long; Lewis J. Rubin; Frank W. Smart; et al.Yuichiro J. SuzukiMark GladwinElizabeth M. DenholmRothy B. Gail Right Ventricular Function and Failure. Circulation 2006, 114, 1883-1891, 10.1161/circulationaha.106.632208.
- Ruxandra Jurcuț; Kristina Haugaa; André La Gerche; The Right Ventricle: From Bench to Bedside. BioMed Research International 2018, 2018, 1-3, 10.1155/2018/2868437.
- Baptiste Rode; Marc A Bailey; Roger Marthan; David J. Beech; Christelle Guibert; ORAI Channels as Potential Therapeutic Targets in Pulmonary Hypertension. Physiology 2018, 33, 261-268, 10.1152/physiol.00016.2018.
- Tian-Tian Zhu; Wei-Fang Zhang; Ping Luo; Zhao-Xin Qian; Feng Li; Zheng Zhang; Changping Hu; LOX-1 promotes right ventricular hypertrophy in hypoxia-exposed rats. Life Sciences 2017, 174, 35-42, 10.1016/j.lfs.2017.02.016.
- Sandro Malacrida; Alessandra Giannella; Giulio Ceolotto; Carlo Reggiani; Alessandra Vezzoli; Simona Mrakic-Sposta; Sarah Moretti; Rachel Turner; Marika Falla; Hermann Brugger; et al.Giacomo Strapazzon Transcription Factors Regulation in Human Peripheral White Blood Cells during Hypobaric Hypoxia Exposure: an in-vivo experimental study. Scientific Reports 2019, 9, 9901, 10.1038/s41598-019-46391-6.
- Yibin Wang; Mitogen-activated protein kinases in heart development and diseases.. Circulation 2007, 116, 1413-23, 10.1161/CIRCULATIONAHA.106.679589.
- R. Dale Brown; S. Kelly Ambler; Min Li; Timothy M. Sullivan; Lauren N. Henry; Joseph T. Crossno; Carlin S. Long; Timothy P. Garrington; Kurt R. Stenmark; MAP kinase kinase kinase-2 (MEKK2) regulates hypertrophic remodeling of the right ventricle in hypoxia-induced pulmonary hypertension.. American Journal of Physiology-Heart and Circulatory Physiology 2013, 304, H269-81, 10.1152/ajpheart.00158.2012.
- Laurence Dewachter; Céline Dewachter; Inflammation in Right Ventricular Failure: Does It Matter?. Frontiers in Physiology 2018, 9, 1056, 10.3389/fphys.2018.01056.
- Yow Keat Tham; Bianca C. Bernardo; Jenny Y. Y. Ooi; Kate L. Weeks; Julie R. McMullen; Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Archives of Toxicology 2015, 89, 1401-1438, 10.1007/s00204-015-1477-x.
- Tsung-I Chen; Yu-Cheng Hsu; Chih-Feng Lien; Jian-Hong Lin; Hung-Wen Chiu; Kun-Ta Yang; Non-Lethal Levels of Oxidative Stress in Response to Short-Term Intermittent Hypoxia Enhance Ca2+Handling in Neonatal Rat Cardiomyocytes. Cellular Physiology and Biochemistry 2014, 33, 513-527, 10.1159/000358631.
- Joseph R. Burgoyne; Heloise Mongue Din; Philip Eaton; Ajay M Shah; Response to "Detailed aspects of redox signaling in cardiac physiology and pathology".. Circulation Research 2013, 112, e2-e2, 10.1161/circresaha.111.300431.
- Jessica N. Peoples; Anita Saraf; Nasab Ghazal; Tyler T. Pham; Jennifer Q. Kwong; Mitochondrial dysfunction and oxidative stress in heart disease. Experimental & Molecular Medicine 2019, 51, 1-13, 10.1038/s12276-019-0355-7.
- Hajime Morimoto; Masafumi Takahashi; Role of Monocyte Chemoattractant Protein-1 in Myocardial Infarction. International journal of biomedical science : IJBS 2007, 3, 159-167.
- Hiroyuki Tsutsui; Shintaro Kinugawa; Shouji Matsushima; Oxidative stress and heart failure. American Journal of Physiology-Heart and Circulatory Physiology 2011, 301, H2181-H2190, 10.1152/ajpheart.00554.2011.
- Chi Zhang; Fan Wang; Yingxia Zhang; Yimin Kang; Haisheng Wang; Mingming Si; Liping Su; Xue Xin; Feng Xue; Fei Hao; et al.Lechu YuJinzhong XuYanlong LiuMingming Xue Celecoxib prevents pressure overload-induced cardiac hypertrophy and dysfunction by inhibiting inflammation, apoptosis and oxidative stress. Journal of Cellular and Molecular Medicine 2015, 20, 116-127, 10.1111/jcmm.12709.
- Subir Kumar Maulik; Santosh Kumar; Oxidative stress and cardiac hypertrophy: a review. Toxicology Mechanisms and Methods 2012, 22, 359-366, 10.3109/15376516.2012.666650.
- Akylbek Sydykov; Argen Mamazhakypov; Aleksandar Petrović; Djuro Kosanovic; Akpay S. Sarybaev; Norbert Weissmann; Hossein Ardeschir Ghofrani; Ralph T. Schermuly; Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Frontiers in Physiology 2018, 9, 609, 10.3389/fphys.2018.00609.
- Nicole Lüneburg; Patricia Siques; Julio Brito; Karem Arriaza; Eduardo Pena; Hans Klose; Fabiola Leon-Velarde; Rainer H. Böger; Long-Term Chronic Intermittent Hypobaric Hypoxia in Rats Causes an Imbalance in the Asymmetric Dimethylarginine/Nitric Oxide Pathway and ROS Activity: A Possible Synergistic Mechanism for Altitude Pulmonary Hypertension?. Pulmonary Medicine 2015, 2016, 1-9, 10.1155/2016/6578578.
- IC Chiş; D Baltaru; A Dumitrovici; A Coseriu; Bc Radu; R Moldovan; A Mureşan; Protective effects of quercetin from oxidative/nitrosative stress under intermittent hypobaric hypoxia exposure in the rat’s heart. Physiology International 2018, 105, 233-246, 10.1556/2060.105.2018.3.23.
- Alejandro Gonzalez-Candia; Marcelino Veliz; Catalina Carrasco-Pozo; Rodrigo L. Castillo; J. Cesar Cárdenas; Germán Ebensperger; Roberto V. Reyes; Aníbal J. Llanos; Emilio A. Herrera; Antenatal melatonin modulates an enhanced antioxidant/pro-oxidant ratio in pulmonary hypertensive newborn sheep. Redox Biology 2019, 22, 101128, 10.1016/j.redox.2019.101128.
- Jayashree Karar; Karamjit S. Dolt; Manoj K. Mishra; Ehtesham Arif; Saleem Javed; M.A. Qadar Pasha; Expression and functional activity of pro-oxidants and antioxidants in murine heart exposed to acute hypobaric hypoxia. FEBS Letters 2007, 581, 4577-4582, 10.1016/j.febslet.2007.08.044.
- Bing Zhang; Ping Zhang; Yanzhen Tan; Pan Feng; Zhengbin Zhang; Hongliang Liang; Weixun Duan; Zhenxiao Jin; Xiaowu Wang; Jincheng Liu; et al.Erhe GaoShiqiang YuDinghua YiYang SunWei Yi C1q-TNF-related protein-3 attenuates pressure overload-induced cardiac hypertrophy by suppressing the p38/CREB pathway and p38-induced ER stress.. Cell Death & Disease 2019, 10, 520-19, 10.1038/s41419-019-1749-0.
- Nurbubu T. Moldogazieva; Innokenty M. Mokhosoev; Nataliya Feldman; Sergey Lutsenko; ROS and RNS signalling: adaptive redox switches through oxidative/nitrosative protein modifications. Free Radical Research 2018, 52, 507-543, 10.1080/10715762.2018.1457217.
- Yi Wen; Ruohong Liu; Ning Lin; Hao Luo; Jiajia Tang; Qilin Huang; Hongyu Sun; Lijun Tang; NADPH Oxidase Hyperactivity Contributes to Cardiac Dysfunction and Apoptosis in Rats with Severe Experimental Pancreatitis through ROS-Mediated MAPK Signaling Pathway.. Oxidative Medicine and Cellular Longevity 2019, 2019, 4578175-18, 10.1155/2019/4578175.
- Ryuichi Aikawa; Toshio Nagai; Mariko Tanaka; Yunzeng Zou; Toshimitsu Ishihara; Hiroyuki Takano; Hiroshi Hasegawa; Hiroshi Akazawa; Miho Mizukami; Ryozo Nagai; et al.Issei Komuro Reactive Oxygen Species in Mechanical Stress-Induced Cardiac Hypertrophy. Biochemical and Biophysical Research Communications 2001, 289, 901-907, 10.1006/bbrc.2001.6068.
- Victoria C. Tu; Joe J. Bahl; Qin M. Chen; Distinct roles of p42/p44(ERK) and p38 MAPK in oxidant-induced AP-1 activation and cardiomyocyte hypertrophy.. Cardiovascular Toxicology 2002, 3, 119-134, 10.1385/ct:3:2:119.
- M Strnisková; T Ravingerová; J Neckár; F Kolár; S Pastoreková; M Barancík; Changes in the expression and/or activation of regulatory proteins in rat hearts adapted to chronic hypoxia.. General physiology and biophysics 2006, 25, 25-41.
- Shih-Wei Hsu; Tsu-Chung Chang; Yu-Kuan Wu; Kuen-Tze Lin; Li-Shian Shi; Shih-Yu Lee; Rhodiola crenulata extract counteracts the effect of hypobaric hypoxia in rat heart via redirection of the nitric oxide and arginase 1 pathway.. BMC Complementary and Alternative Medicine 2017, 17, 29, 10.1186/s12906-016-1524-z.
- S. Kumar; S. M. Blake; Pharmacological Potential of p38 MAPK Inhibitors. Handbook of Experimental Pharmacology 2004, 167, 65-83, 10.1007/3-540-26670-4_4.
- Bo Yu; Yanan Zhao; Hongli Zhang; Di Xie; Wei Nie; Kaiyao Shi; Inhibition of microRNA-143-3p attenuates myocardial hypertrophy by inhibiting inflammatory response.. Cell Biology International 2018, 42, 1584-1593, 10.1002/cbin.11053.
- Alexandre Sarre; Norbert Lange; Pavel Kucera; Eric Raddatz; mitoKATP channel activation in the postanoxic developing heart protects E-C coupling via NO-, ROS-, and PKC-dependent pathways. American Journal of Physiology-Heart and Circulatory Physiology 2005, 288, H1611-H1619, 10.1152/ajpheart.00942.2004.
- M. Uenoyama; S. Ogata; K. Nakanishi; F. Kanazawa; S. Hiroi; S. Tominaga; A. Seo; T. Matsui; T. Kawai; S. Suzuki; et al. Protein kinase C mRNA and protein expressions in hypobaric hypoxia-induced cardiac hypertrophy in rats. Acta Physiologica 2010, 198, 431-440, 10.1111/j.1748-1716.2009.02064.x.
- Robert E. Carraway; Paul R. Dobner; Zinc pyrithione induces ERK- and PKC-dependent necrosis distinct from TPEN-induced apoptosis in prostate cancer cells. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2012, 1823, 544-557, 10.1016/j.bbamcr.2011.09.013.
- Andrey V. Krylatov; Leonid N. Maslov; Nikita Voronkov; A. A. Boshchenko; Sergey V. Popov; Ludovic Gomez; Hongxin Wang; Amteshwar S. Jaggi; James M. Downey; Reactive Oxygen Species as Intracellular Signaling Molecules in the Cardiovascular System. Current Cardiology Reviews 2018, 14, 290-300, 10.2174/1573403x14666180702152436.
- Xiang Song; Xiaoqian Qian; Ming Shen; Rong Jiang; Mary B. Wagner; Guoliang Ding; Guangping Chen; Baozhong Shen; Protein kinase C promotes cardiac fibrosis and heart failure by modulating galectin-3 expression. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2015, 1853, 513-521, 10.1016/j.bbamcr.2014.12.001.
- Abhinav Diwan; Gerald W. Dorn; Decompensation of Cardiac Hypertrophy: Cellular Mechanisms and Novel Therapeutic Targets. Physiology 2007, 22, 56-64, 10.1152/physiol.00033.2006.
- Zhancui Dang; Shanshan Su; Guoen Jin; Xingmei Nan; Lan Ma; Zhanqiang Li; Dianxiang Lu; Ri-Li Ge; Tsantan Sumtang attenuated chronic hypoxia-induced right ventricular structure remodeling and fibrosis by equilibrating local ACE-AngII-AT1R/ACE2-Ang1-7-Mas axis in rat. Journal of Ethnopharmacology 2020, 250, 112470, 10.1016/j.jep.2019.112470.
- B. Kojonazarov; Tatyana Novoyatleva; Mario Boehm; Chris M. Happé; Zaneta Sibinska; Xia Tian; Amna Sajjad; Himal Luitel; Philipp Kriechling; Guido Posern; et al.Steven M. EvansFriedrich GrimmingerHossein Ardeschir GhofraniNorbert WeissmannHarm J. BogaardWerner SeegerRalph T. Schermuly p38 MAPK Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. American Journal of Respiratory Cell and Molecular Biology 2017, 57, 603-614, 10.1165/rcmb.2016-0374oc.
- Karen E. Porter; Neil A. Turner; Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacology & Therapeutics 2009, 123, 255-278, 10.1016/j.pharmthera.2009.05.002.
- Jia-Hong Wang; Lan Zhao; Xin Pan; Nan-Nan Chen; Jian Chen; Qun-Lin Gong; Feng Su; Jian Yan; Yan Zhang; Shao-Heng Zhang; et al. Erratum: Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-β1 signaling pathway. Laboratory Investigation 2016, 96, 1035-1035, 10.1038/labinvest.2016.84.
- Jennifer Baum; Heather S. Duffy; Fibroblasts and Myofibroblasts: What Are We Talking About?. Journal of Cardiovascular Pharmacology 2011, 57, 376-379, 10.1097/fjc.0b013e3182116e39.
- Maarten Hulsmans; Flora Sam; Matthias Nahrendorf; Monocyte and macrophage contributions to cardiac remodeling. Journal of Molecular and Cellular Cardiology 2016, 93, 149-155, 10.1016/j.yjmcc.2015.11.015.
- Havovi Chichger; Alexander Vang; Kelly A. O'connell; Peng Zhang; Ulrike Mende; Elizabeth O. Harrington; Gaurav Choudhary; PKC δ and βII regulate angiotensin II-mediated fibrosis through p38: a mechanism of RV fibrosis in pulmonary hypertension.. American Journal of Physiology-Lung Cellular and Molecular Physiology 2015, 308, L827-36, 10.1152/ajplung.00184.2014.
- Maulik D. Majmudar; Edmund J. Keliher; Timo Heidt; Florian Leuschner; Jessica Truelove; Brena F. Sena; Rostic Gorbatov; Yoshiko Iwamoto; Partha Dutta; Gregory R. Wojtkiewicz; et al.Gabriel CourtiesMatt SebasAnna BorodovskyKevin FitzgeraldMarc W. NolteGerhard DickneiteJohn W. ChenDaniel G. AndersonFilip K. SwirskiRalph WeisslederMatthias Nahrendorf Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013, 127, 2038-2046, 10.1161/CIRCULATIONAHA.112.000116.
- Anthony S. Jaipersad; Gregory Y.H. Lip; Stanley Silverman; Eduard Shantsila; The Role of Monocytes in Angiogenesis and Atherosclerosis. Journal of the American College of Cardiology 2014, 63, 1-11, 10.1016/j.jacc.2013.09.019.
- Santosh Kumar; Gang Wang; Na Zheng; Wanwen Cheng; Kunfu Ouyang; Hairuo Lin; Yulin Liao; Jie Liu; HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis.. Hypertension 2019, 73, 1058-1070, 10.1161/HYPERTENSIONAHA.118.12267.
- Linlin Jing; Jin Shao; Wei Sun; Ting Lan; ZhengPing Jia; Huiping Ma; Haibo Wang; Protective effects of two novel nitronyl nitroxide radicals on heart failure induced by hypobaric hypoxia. Life Sciences 2020, 248, 116481, 10.1016/j.lfs.2019.05.037.
- Harm J. Bogaard; Kohtaro Abe; Anton Vonk Noordegraaf; Norbert F. Voelkel; The Right Ventricle Under Pressure. Chest 2009, 135, 794-804, 10.1378/chest.08-0492.
- Antonino Nicoletti; J B Michel; Cardiac fibrosis and inflammation: interaction with hemodynamic and hormonal factors.. Cardiovascular Research 1999, 41, 532-543, 10.1016/S0008-6363(98)00305-8.
- Yun-Ching Fu; Ching-Shiang Chi; Sui-Chu Yin; Betau Hwang; Yung-Tsung Chiu; Shih-Lan Hsu; Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species?TNF$alpha;?caspase signaling pathway. Cardiovascular Research 2004, 62, 558-567, 10.1016/j.cardiores.2004.01.039.
- Xinyu Li; Quyan Zhang; M I Nasser; Linyong Xu; Xueyan Zhang; Ping Zhu; Qingnan He; Mingyi Zhao; Oxygen homeostasis and cardiovascular disease: A role for HIF?. Biomedicine & Pharmacotherapy 2020, 128, 110338, 10.1016/j.biopha.2020.110338.
- Deepak Bhatia; Mohammad Sanaei Ardekani; Qiwen Shi And Shahrzad Movafagh; Hypoxia and its Emerging Therapeutics in Neurodegenerative, Inflammatory and Renal Diseases. Hypoxia and Human Diseases 2017, 1, 404-443, 10.5772/66089.
- Sarita Nehra; Varun Bhardwaj; Anju Bansal; Deepika Saraswat; Combinatorial therapy of exercise-preconditioning and nanocurcumin formulation supplementation improves cardiac adaptation under hypobaric hypoxia. Journal of Basic and Clinical Physiology and Pharmacology 2016, 28, 443-453, 10.1515/jbcpp-2016-0134.
- Manimaran, Manickam; RajkumarTulsawani; Duraisamy Arul, Joseph; Vishal, Jain; Kshipra, Misra; Protective efficacy of Hippophae rhamnoides L. extract exhibited in rat heart against hypobaric hypoxia is possibly mediated by configurations in JAK/STAT pathway. Indian J. Exp. Biol. 2019, 57, 390-398, http://nopr.niscair.res.in/handle/123456789/47442.
- Qingshu Wang; Ling Hu; Yu Hu; Gu Gong; Hua Tan; Li Deng; Xiaoqin Sun; Xiaobo Yi; Yangyang Sun; Wei Wu; et al.Tao Li Carbon Monoxide-Saturated Hemoglobin-Based Oxygen Carriers Attenuate High-Altitude-Induced Cardiac Injury by Amelioration of the Inflammation Response and Mitochondrial Oxidative Damage. Cardiology 2016, 136, 180-191, 10.1159/000448652.
- Yun-Yun He; Chun-Lei Liu; Xin Li; Rui-Jun Li; Li-Li Wang; Kun-Lun He; Salubrinal attenuates right ventricular hypertrophy and dysfunction in hypoxic pulmonary hypertension of rats. Vascular Pharmacology 2016, 87, 190-198, 10.1016/j.vph.2016.09.009.
- Yali Wang; Feng Wu; Feng Hu; Yunjiang Wu; Jun Zhou; Yan Xu; Xiangrong Shao; Tao Hu; Drag-reducing polymers attenuates pulmonary vascular remodeling and right ventricular dysfunction in a rat model of chronic hypoxia-induced pulmonary hypertension. Clinical Hemorheology and Microcirculation 2020, 74, 189-200, 10.3233/ch-190668.
- Jie Ren; Wei Liu; Guang-Cai Li; M. Jin; Zhen-Xi You; Hui-Guo Liu; Yi Hu; Atorvastatin Attenuates Myocardial Hypertrophy Induced by Chronic Intermittent Hypoxia In Vitro Partly through miR-31/PKCε Pathway. Current Medical Science 2018, 38, 405-412, 10.1007/s11596-018-1893-2.

