 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michele Simbolo | + 2154 word(s) | 2154 | 2022-02-16 05:18:07 | | | |
| 2 | Amina Yu | -2 word(s) | 2152 | 2022-02-17 02:55:43 | | | | |
| 3 | Amina Yu | Meta information modification | 2152 | 2022-02-18 07:37:23 | | |
Video Upload Options
The use of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) as first-line treatment in patients with lung adenocarcinoma (LUAD) harboring EGFR-activating mutations has resulted in a dramatic improvement in the management of the disease. Lung cancer remains one of the most frequent and deadly types of cancer for both genders, while predictions about the future do not suggest dramatic improvements in incidence and mortality rates. There are different histological types of primary lung cancer, classified mainly into non-small-cell lung cancer (NSCLC) and small-cell lung cancer, with an annual incidence of approximately 85% and 15%, respectively. Lung adenocarcinoma (LUAD) is the most common subtype of NSCLC, accounting for around 40% of NSCLC cases.
1. Introduction
2. EGFR-Mutant LUAD Global Metabolic Profile
3. EGFR TKI Resistance and Glycolysis/Lactate Metabolism
4. EGFR TKI Resistance and Fatty Acid Metabolism
5. EGFR TKI Resistance and Redox Homeostasis
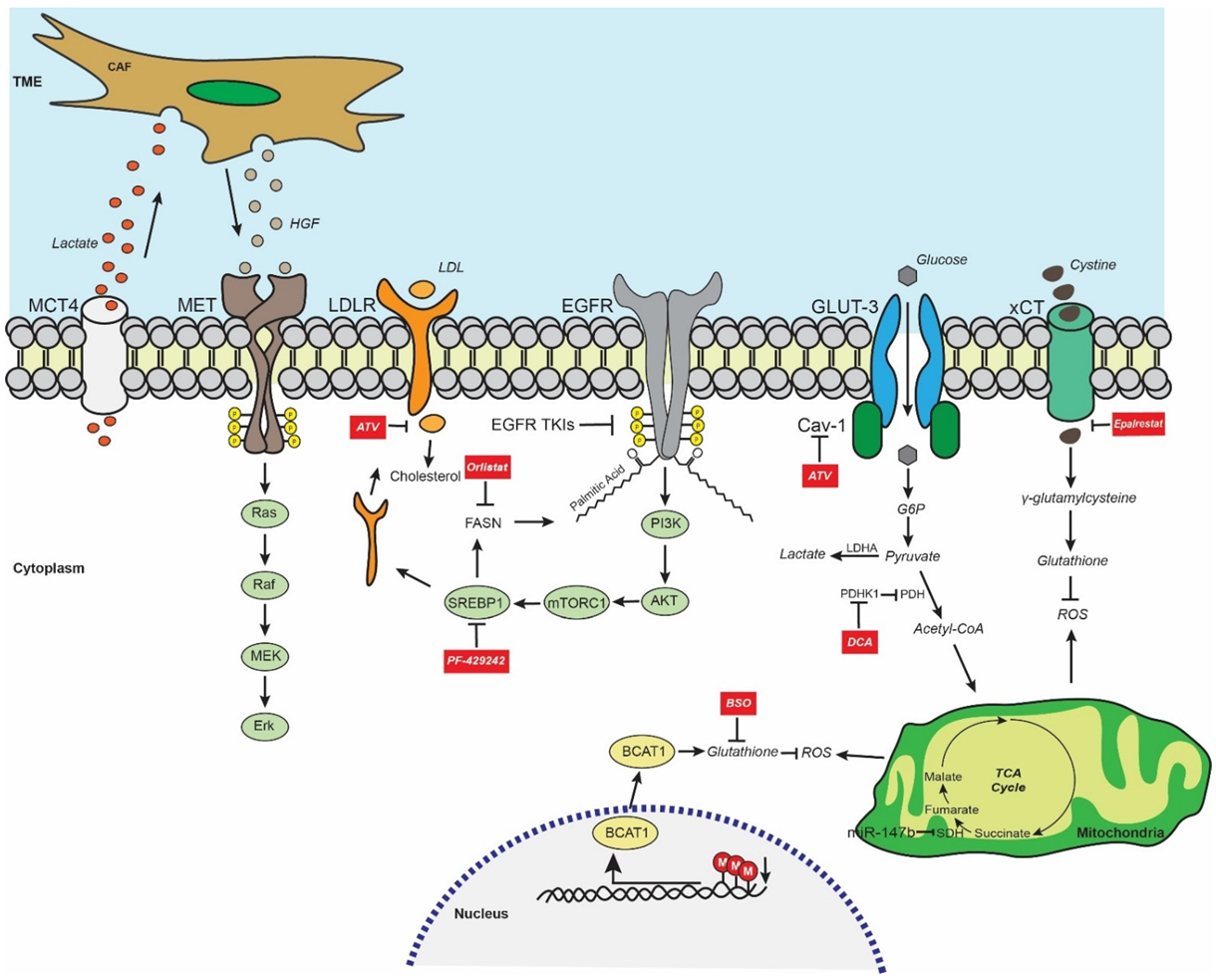
6. EGFR TKI Resistance and Metabolism-Mediated Stemness
References
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957.
- Douillard, J.Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; McCormack, R.; Webster, A.; Milenkova, T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: A phase-IV, open-label, single-arm study. Br. J. Cancer 2014, 110, 55–62.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742.
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151.
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38.
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247.
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640.
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50.
- Papadimitrakopoulou, V.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.C.H.; Bulusu, K.; Laus, G.; et al. LBA51Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741.
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534.
- Schmid, S.; Li, J.J.N.; Leighl, N.B. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer 2020, 147, 123–129.
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation 2021, 2, 100103.
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52.
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88.
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240.
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823.
- Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.N.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.T.; Daubon, T.; Mourier, A.; Thiery, J.P.; et al. Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 941.
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e6.
- Han, X.; Luo, R.; Wang, L.; Zhang, L.; Wang, T.; Zhao, Y.; Xiao, S.; Qiao, N.; Xu, C.; Ding, L.; et al. Potential predictive value of serum targeted metabolites and concurrently mutated genes for EGFR-TKI therapeutic efficacy in lung adenocarcinoma patients with EGFR sensitizing mutations. Am. J. Cancer Res. 2020, 10, 4266–4286.
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248.
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137.
- Nguyen, P.A.; Chang, C.C.; Galvin, C.J.; Wang, Y.C.; An, S.Y.; Huang, C.W.; Wang, Y.H.; Hsu, M.H.; Li, Y.J.; Yang, H.C. Statins use and its impact in EGFR-TKIs resistance to prolong the survival of lung cancer patients: A Cancer registry cohort study in Taiwan. Cancer Sci. 2020, 111, 2965–2973.
- Ali, A.; Levantini, E.; Fhu, C.W.; Teo, J.T.; Clohessy, J.G.; Goggi, J.L.; Wu, C.S.; Chen, L.; Chin, T.M.; Tenen, D.G. CAV—GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics 2019, 9, 6157–6174.
- Martinez, C.A.; Scafoglio, C. Heterogeneity of Glucose Transport in Lung Cancer. Biomolecules 2020, 10, 868.
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11.
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176.
- Jin, N.; Bi, A.; Lan, X.; Xu, J.; Wang, X.; Liu, Y.; Wang, T.; Tang, S.; Zeng, H.; Chen, Z.; et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat. Commun. 2019, 10, 2701.
- Zhang, K.R.; Zhang, Y.F.; Lei, H.M.; Tang, Y.B.; Ma, C.S.; Lv, Q.M.; Wang, S.Y.; Lu, L.M.; Shen, Y.; Chen, H.Z.; et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci. Transl. Med. 2021, 13, eabg6428.
- Hashida, S.; Yamamoto, H.; Shien, K.; Miyoshi, Y.; Ohtsuka, T.; Suzawa, K.; Watanabe, M.; Maki, Y.; Soh, J.; Asano, H.; et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015, 106, 1377–1384.
- Jia, Z.; Zhang, Y.; Yan, A.; Wang, M.; Han, Q.; Wang, K.; Wang, J.; Qiao, C.; Pan, Z.; Chen, C.; et al. 1,25-dihydroxyvitamin D3 signaling-induced decreases in IRX4 inhibits NANOG-mediated cancer stem-like properties and gefitinib resistance in NSCLC cells. Cell Death Dis. 2020, 11, 670.
- Ji, M.; Liu, L.; Hou, Y.; Li, B. 1alpha,25Dihydroxyvitamin D3 restrains stem celllike properties of ovarian cancer cells by enhancing vitamin D receptor and suppressing CD44. Oncol. Rep. 2019, 41, 3393–3403.
- Polonio, E.; Palomeras, S.; Porta-Balanya, R.; Bosch-Barrera, J.; Vásquez, C.A.; Ciurana, J.; Ruiz-Martínez, S.; Puig, T. 3P AZ12756122, a novel fatty acid synthase (FASN) inhibitor, reduces resistance properties in gefitinib- and osimertinib-resistant EGFR-mutated non-small cell lung cancer models. J. Thorac. Oncol. 2021, 16, S700.

