Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marina Stogniy | + 1058 word(s) | 1058 | 2022-02-09 10:17:44 | | | |
| 2 | Rita Xu | Meta information modification | 1058 | 2022-02-10 02:48:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Stogniy, M. Oxonium Derivatives of nido-Carborane. Encyclopedia. Available online: https://encyclopedia.pub/entry/19278 (accessed on 23 July 2026).
Stogniy M. Oxonium Derivatives of nido-Carborane. Encyclopedia. Available at: https://encyclopedia.pub/entry/19278. Accessed July 23, 2026.
Stogniy, Marina. "Oxonium Derivatives of nido-Carborane" Encyclopedia, https://encyclopedia.pub/entry/19278 (accessed July 23, 2026).
Stogniy, M. (2022, February 09). Oxonium Derivatives of nido-Carborane. In Encyclopedia. https://encyclopedia.pub/entry/19278
Stogniy, Marina. "Oxonium Derivatives of nido-Carborane." Encyclopedia. Web. 09 February, 2022.
Copy Citation
Recent decades have demonstrated a growing interest in the chemistry of 7,8-dicarba-nido-undecaborante anion (nido-carborane) due to the wide possibilities of its application from medicine to catalysis. One of the main approaches to the modification of nido-carborane cluster is the ring-opening reactions of its cyclic oxonium derivatives with various nucleophiles, which opens practically unlimited prospects for the incorporation of nido-carborane into various macro- and biomolecules.
nido-carborane
half-sandwich complexes
oxonium derivatives
ring-opening reactions
1. Introduction
This anionic boron cluster with an open pentagonal face was synthesized for the first time by M. F. Hawthorne more than 50 years ago by deboronation of 1,2-dicarba-closo-dodecaborane (ortho-carborane) with potassium hydroxide in methanol [1][2]. Later various milder deboronating agents including amines [3][4][5][6] and fluoride ion [7][8][9][10] were proposed. Deprotonation of nido-carborane with strong bases results in the dicarbollide dianion [7,8-C2B9H11]2−, which was recognized as an isolobal analogue of the cyclopentadienyl anion [11]. Due to this, the dicarbollide dianion is considered as an unusual three-dimensional π-ligand for the synthesis of metal complexes [12][13][14], which can be used as catalysts in a variety of chemical processes [15][16]. Another important area of use for nido-carborane derivatives as water-solubilizing boron moieties is the design of potential drugs for boron neutron capture therapy of cancer [17][18][19][20][21][22]. In addition, based on nido-carborane derivatives, it is possible to obtain reagents for radioimaging of tumors [23][24][25][26][27], new optical and luminescent materials [28][29][30][31][32][33][34][35][36], ionic liquids [37][38], etc. Thus, the development of convenient approaches to the modification of the nido-carborane cluster remains highly relevant.
Currently the main approach to the synthesis of nido-carborane derivatives is based on the deboronation of the corresponding ortho-carborane derivatives. This approach is widely used for the synthesis of C-substituted derivatives of nido-carborane, as well as derivatives containing substituents at the lower belt of the nido-carborane cage [39]. There are several general methods for the synthesis of B-substituted derivatives with substituents in the upper belt of the nido-carborane cage; however, most of them are used to obtain asymmetrically substituted derivatives [9-X-7,8-C2B9H11]− [40][41][42][43][44][45][46][47][48].
Several approaches to the synthesis of symmetrically substituted derivatives of nido-carborane [10-X-7,8-C2B9H11]− with boron–sulfur [49][50] and boron–nitrogen [51][52][53][54] bonds have also been developed recently; however, the greatest interest is the functionalization of nido-carborane via its cyclic oxonium derivatives. Previously, this approach was successfully used for the synthesis of numerous derivatives of closo-decaborate and closo-dodecaborate anions [55][56], as well as sandwich bis(dicarbollide) transition metal complexes [57].
2. Synthesis of Oxonium Derivatives of nido-Carborane
To date, oxonium derivatives of nido-carborane have been synthesized with all cyclic ethers used as common solvents, including tetrahydrofuran, 1,4-dioxane, and tetrahydropyran. The first oxonium derivative was obtained as early as 1969 by the reaction of the potassium salt of nido-carborane with FeCl3 in a mixture of benzene and tetrahydrofuran. The reaction gave a mixture of asymmetric 9-(CH2)4O-7,8-C2B9H11 and symmetric 10-(CH2)4O-7,8-C2B9H11 which were separated by column chromatography in benzene. Similar mixture of isomers was obtained for the C,C-dimethyl derivative of nido-carborane as well [58] (Scheme 1).
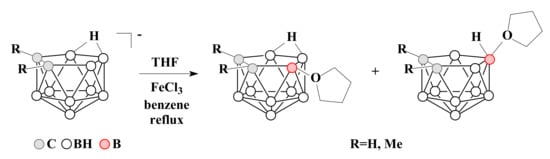
Scheme 1. The synthesis of 9-(CH2)4O-7,8-C2B9H11 and 10-(CH2)4O-7,8-C2B9H11 by the reaction of nido-carborane with FeCl3 in a THF–benzene mixture.
The proposed mechanism of this reaction includes the initial abstraction of hydrogen hydride from position 9 by iron(III) chloride as a Lewis acid with the formation of a quasi-borinium cation. In the absence of strong nucleophiles, this intermediate can be isomerized to a more stable symmetric form with a quasi-electrophilic center at position 10, which is then attacked by an ether solvent molecule as a weak but most accessible nucleophile. As a result, a mixture of 9- and 10-substituted isomers is formed [59]. This mechanism is known as electrophile-induced nucleophilic substitution (EINS) and is considered as one of the main mechanisms of substitution of hydrogen atoms in polyhedral boron hydrides.
Later, it was found that the reaction of nido-carborane with HgCl2 in tetrahydrofuran leads to the selective formation of the symmetrically substituted product 10-(CH2)4O-7,8-C2B9H11 [59][60]. In a similar way, the derivatives with 1,4-dioxane 10-O(CH2CH2)2O-7,8-C2B9H11 [59] and tetrahydropyran 10-(CH2)5O-7,8-C2B9H11 [61][62] were synthesized.
It is assumed that the selectivity of the reactions in these cases is determined by the fact that the endo-polyhedral hydrogen atom of nido-carborane cage is replaced at the first stage of the reaction by a mercury atom with the formation of η1-metallacarborane, in which mercury is bound to the B(10) atom of the dicarbollide ligand [63][64][65][66]. Heating of this complex results in elimination of mercury and generation of quasi-electrophilic center at position 10 followed by its attack by the nucleophile [59].
Another convenient method for the synthesis of 1,4-dioxane derivative of nido-carborane is to heat in 1,4-dioxane the protonated form of nido-carborane 7,8-C2B9H13, which is formed by treating a suspension of the triethylammonium salt of nido-carborane with concentrated sulfuric acid in toluene [67].
The symmetrically substituted tetrahydrofuran derivative 10-(CH2)4O-7,8-C2B9H11 can be obtained by the reaction of tetramethylammonium salt of nido-carborane with AlCl3 in a mixture of tetrahydrofuran and acetone [68]. The symmetrically substituted tetrahydrofuran and 1,4-dioxane derivatives of nido-carborane were also synthesized by the reactions of nido-carborane with the corresponding ethers in the presence of acetaldehyde or formaldehyde and hydrochloric acid in a mixture of water and toluene [69]. The methods of synthesis of symmetrically substituted oxonium derivatives of nido-carborane are summarized in Scheme 2.
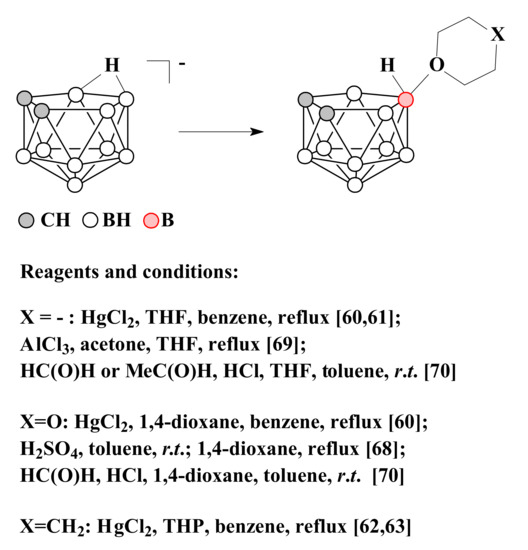
Scheme 2. The different synthetic pathways to the 10-(CH2)4O-7,8-C2B9H11, 10-O(CH2CH2)2O-7,8-C2B9H11 and 10-(CH2)5O-7,8-C2B9H11.
The molecular structure of the 1,4-dioxane derivative of nido-carborane was determined by single crystal X-ray diffraction [70] (Figure 1).
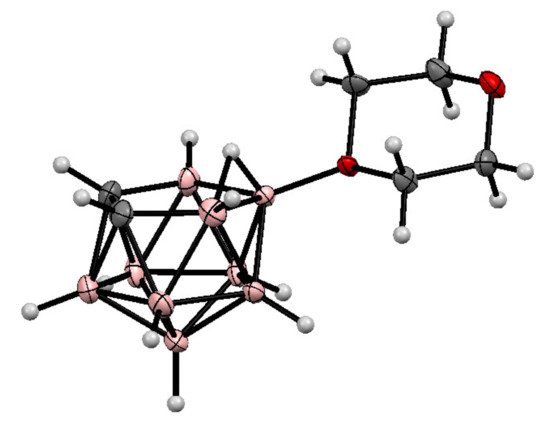
Figure 1. The molecular structure of 10-O(CH2CH2)2O-7,8-C2B9H11.
It should be noted that, in addition to cyclic oxonium derivatives of nido-carborane, some acyclic oxonium derivatives such as the dimethyloxonium 9-Me2O-7,8-C2B9H11 and 10-Me2O-7,8-C2B9H11 [71] and the diethyloxonium 9-Et2O-7,8-C2B9H11 [72] and 10-Et2O-7,8-C2B9H11 [69][72][73] derivatives are known as well. The molecular structure of the 10-diethyloxonium derivative of nido-carborane was determined by single crystal X-ray diffraction [72] (Figure 2). The formation of the 1,2-dimethoxyethane derivatives 9-MeOCH2CH2(Me)O-7,8-C2B9H11 and 10-MeOCH2CH2(Me)O-7,8-C2B9H11 was also reported, however they have low stability and easily lose the methyl group, giving the corresponding alkoxy derivatives [74].
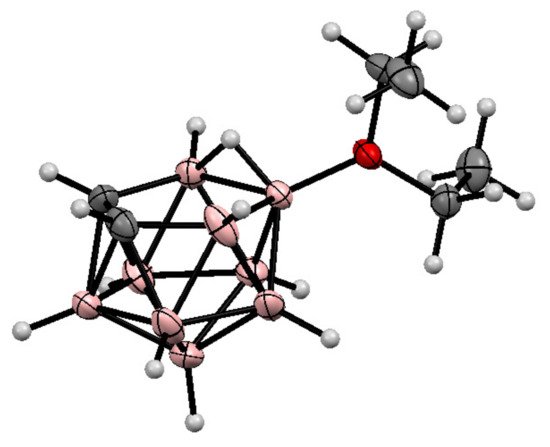
Figure 2. The molecular structure of 10-Et2O-7,8-C2B9H11.
References
- Wiesboeck, R.A.; Hawthorne, M.F. Dicarbaundecaborane(13) and Derivatives. J. Am. Chem. Soc. 1964, 86, 1642–1643.
- Hawthorne, M.F.; Young, D.C.; Garrett, P.M.; Owen, D.A.; Schwerin, S.G.; Tebbe, F.N.; Wegner, P.A. Preparation and characterization of the (3)-1,2- and (3)-1,7-dicarbadodecahydroundecarborate(-1) ions. J. Am. Chem. Soc. 1968, 90, 862–868.
- Zakharkin, L.I.; Kalinin, V.I. On the reaction of amines with barenes. Tetrahedron Lett. 1965, 7, 407–409.
- Hawthorne, M.F.; Wegner, P.A.; Stafford, R.C. Comments on the reaction of amines with 1,2-dicarbaclovododecaborane(12). Inorg. Chem. 1965, 4, 1675.
- Davidson, M.G.; Fox, M.A.; Hibbert, T.G.; Howard, J.A.K.; Mackinnon, A.; Neretin, I.S.; Wade, K. Deboronation of ortho-carborane by an iminophosphorane: Crystal structures of the novel carborane adduct nido-C2B10H12·HNP(NMe2)3 and the borenium salt 2O2+(C2B9H12−)2. Chem. Commun. 1999, 1649–1650.
- Wise, S.D.; Au, W.; Getman, T.D. The effect of varying the solvent system on the rate of the deboration of o-carborane by n-butyl amine. Main Group Met. Chem. 2002, 25, 411–413.
- Tomita, H.; Luu, H.; Onak, T. Cage opening of parent closo cage carboranes with fluoride ion: Formation of 5-fluoro-hexahydro-nido-2,4-dicarbahexaborate(1-) (−). Inorg. Chem. 1991, 30, 812–815.
- Fox, M.A.; Gill, W.R.; Herbertson, P.L.; MacBride, J.A.H.; Wade, K.; Colquhoun, H.M. Deboronation of C-substituted ortho- and meta-closo-carboranes using “wet” fluoride ion solutions. Polyhedron 1996, 15, 565–571.
- Getman, T.D. Investigation of potassium fluoride supported on alumina in the deboronation of o-carborane. Inorg. Chem. 1998, 37, 3422–3423.
- Yoo, J.; Hwang, J.-W.; Do, Y. Facile and mild deboronation of o-carboranes using cesium fluoride. Inorg. Chem. 2001, 40, 568–570.
- Hawthorne, M.F.; Young, D.C.; Andrews, T.D.; Howe, D.V.; Pilling, R.L.; Pitts, A.D.; Reintjer, M.; Warren, L.F.; Wegner, P.A. π-Dicarbollyl derivatives of the transition metals. Metallocene analogs. J. Am. Chem. Soc. 1968, 90, 879–896.
- Grimes, R.N. Transitional metal metallacarbaboranes. In Comprehensive Organometallic Chemistry II; Elsevier: Oxford, UK, 1995; Volume 1, pp. 373–430.
- Grimes, R.N. Metallacarboranes in the new millennium. Co-ord. Chem. Rev. 2000, 200, 773–811.
- Hosmane, N.S.; Maguire, J.A. Metallacarboranes of d- and f-block metals. In Comprehensive Organometallic Chemistry III; Elsevier: Oxford, UK, 2007; Volume 3, pp. 175–264.
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and mixed-sandwich metallacarboranes in catalysis. In Handbook of Boron Chemistry with Applications in Organometallics, Catalysis, Materials and Medicine. Boron in Catalysis; Hosmane, N.S., Eagling, R., Eds.; World Scientific Publishing Europe Ltd.: London, UK, 2019; Volume 2, pp. 27–80.
- Zhu, Y.; Hosmane, N.S. Carborane-based catalysts for polymerization of olefins. In Handbook of Boron Chemistry with Applications in Organometallics, Catalysis, Materials and Medicine. Boron in Catalysis; Hosmane, N.S., Eagling, R., Eds.; World Scientific Publishing Europe Ltd.: London, UK, 2019; Volume 2, pp. 117–134.
- Hawthorne, M.F. The role of chemistry in the development of boron neutron capture therapy of cancer. Angew. Chem. Int. Ed. Engl. 1993, 32, 950–984.
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The chemistry of neutron capture therapy. Chem. Rev. 1998, 98, 1515–1562.
- Valliant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K.A. The medicinal chemistry of carboranes. Co-ord. Chem. Rev. 2002, 232, 173–230.
- Armstrong, A.F.; Valliant, J.F. The bioinorganic and medicinal chemistry of carboranes: From new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 4240–4251.
- Lee, W.; Sarkar, S.; Ahn, H.; Kim, J.Y.; Lee, Y.J.; Chang, Y.; Yoo, J. PEGylated liposome encapsulating nido-carborane showed significant tumor suppression in boron neutron capture therapy (BNCT). Biochem. Biophys. Res. Commun. 2020, 522, 669–675.
- Varaksin, M.V.; Smyshliaeva, L.A.; Rusinov, V.L.; Makeev, O.G.; Melekhin, V.V.; Baldanshirieva, A.D.; Gubina, O.G.; Charushin, V.N.; Chupakhin, O.N. Synthesis, characterization, and in vitro assessment of cytotoxicity for novel azaheterocyclic nido-carboranes—Candidates in agents for boron neutron capture therapy (BNCT) of cancer. Tetrahedron 2021, 102, 132525.
- Tolmachev, V.V.; Sjöberg, S. Polyhedral boron compounds as potential linkers for attachment of radiohalogens to targeting proteins and peptides. A review. Collect. Czech. Chem. Commun. 2002, 67, 913–935.
- Wilbur, D.S.; Chyan, M.-C.; Hamlin, D.K.; Vessella, R.L.; Wedge, T.J.; Hawthorne, M.F. Reagents for astatination of biomolecules. 2. Conjugation of anionic boron cage pendant groups to a protein provides a method for direct labeling that is stable to in vivo deastatination. Bioconjugate Chem. 2007, 18, 1226–1240.
- Green, A.E.C.; Harrington, L.E.; Valliant, J.F. Carborane-carbohydrate derivatives—Versatile platforms for developing targeted radiopharmaceuticals. Can. J. Chem. 2008, 86, 1063–1069.
- Armstrong, A.F.; Lebert, J.M.; Brennan, J.D.; Valliant, J.F. Functionalized carborane complexes of the 2+ core (M = 99mTc, Re): A new class of organometallic probes for correlated in vitro and in vivo imaging. Organometallics 2009, 28, 2986–2992.
- El-Zaria, M.E.; Janzen, N.; Valliant, J.F. Room-temperature synthesis of Re(I) and Tc(I) metallocarboranes. Organometallics 2012, 31, 5940–5949.
- Visbal, R.; Ospino, I.; López-de-Luzuriaga, J.M.; Laguna, A.; Gimeno, M.C. N-Heterocyclic carbene ligands as modulators of luminescence in three-coordinate gold(I) complexes with spectacular quantum yields. J. Am. Chem. Soc. 2013, 135, 4712–4715.
- Axtell, J.C.; Kirlikovali, K.O.; Djurovich, P.I.; Jung, D.; Nguyen, V.T.; Munekiyo, B.; Royappa, A.T.; Rheingold, A.L.; Spokoyny, A.M. Blue phosphorescent zwitterionic iridium(III) complexes featuring weakly coordinating nido-carborane-based ligands. J. Am. Chem. Soc. 2016, 138, 15758–15765.
- Shafikov, M.Z.; Suleymanova, A.F.; Czerwieniec, R.; Yersin, H. Design strategy for Ag(I)-based thermally activated delayed fluorescence reaching an efficiency breakthrough. Chem. Mater. 2017, 29, 1708–1715.
- Nghia, N.V.; Jana, S.; Sujith, S.; Ryu, J.Y.; Lee, J.; Lee, S.U.; Lee, M.H. nido-Carboranes: Donors for thermally activated delayed fluorescence. Angew. Chem. Int. Ed. 2018, 57, 12483–12488.
- Nghia, N.V.; Oh, J.; Sujith, S.; Jung, J.; Lee, M.H. Tuning the photophysical properties of carboranyl luminophores by closo- to nido-carborane conversion and application to OFF–ON fluoride sensing. Dalton Trans. 2018, 47, 17441–17449.
- He, T.-F.; Ren, A.-M.; Chen, Y.-N.; Hao, X.-L.; Shen, L.; Zhang, B.-H.; Wu, T.-S.; Zhang, H.-X.; Zou, L.-Y. Molecular-level insight of Cu(I) complexes with the 7,8-bis(diphenylphosphino)-7,8-dicarba-nido-undecaborate ligand as a thermally activated delayed fluorescence emitter: Luminescent mechanism and design strategy. Inorg. Chem. 2021, 59, 12039–12053.
- Li, Q.; Shi, C.; Huang, M.; Zhang, X.; Sun, F.; Zheng, Y.; Yan, H.; Yang, C.; Yuan, A. Three types of charged ligand-based neutral phosphorescent iridium(III) complexes featuring nido-carborane: Synthesis, structures, and solution processed organic light-emitting diode applications. Dalton Trans. 2021, 50, 16304–16310.
- Alconchel, A.A.; Crespo, O.; García-Orduña, P.; Gimeno, M.C. closo- or nido-Carborane diphosphane as responsible for strong thermochromism or time activated delayed fluorescence (TADF) in 0. Inorg. Chem. 2021, 60, 18521–18528.
- Lerouge, F.; Ferrer-Ugalde, A.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Abreu, A.; Xochitiotzi, E.; Farfán, N.; Santillan, R.; Núñez, R. Synthesis and fluorescence emission of neutral and anionic di- and tetra-carboranyl compounds. Dalton Trans. 2011, 40, 7541–7550.
- Zhu, Y.; Hosmane, N.S. Ionic liquids: Recent advances and applications in boron chemistry. Eur. J. Inorg. Chem. 2017, 38-39, 4369–4377.
- Sivaev, I.B. Nitrogen heterocyclic salts of polyhedral borane anions: From ionic liquids to energetic materials. Chem. Heterocycl. Compd. 2017, 53, 638–658.
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: London, UK, 2016; pp. 179–257.
- Zakharova, M.V.; Sivaev, I.B.; Anufriev, S.A.; Timofeev, S.V.; Suponitsky, K.Y.; Godovikov, I.A.; Bregadze, V.I. A new approach to the synthesis of functional derivatives of nido-carborane: Alkylation of −. Dalton Trans. 2014, 43, 5044–5053.
- Anufriev, S.A.; Zakharova, M.V.; Sivaev, I.B.; Bregadze, V.I. New carborane-containing acids and amines. Russ. Chem. Bull. 2017, 66, 1643–1649.
- Timofeev, S.V.; Zhidkova, O.B.; Prikaznova, E.A.; Sivaev, I.B.; Semioshkin, A.; Godovikov, I.A.; Starikova, Z.A.; Bregadze, V.I. Direct synthesis of nido-carborane derivatives with pendant functional groups by copper-promoted reactions with dimethylalkylamines. J. Organomet. Chem. 2014, 757, 21–27.
- Druzina, A.A.; Zhidkova, O.B.; Dudarova, N.V.; Kosenko, I.D.; Ananyev, I.V.; Timofeev, S.V.; Bregadze, V.I. Synthesis and structure of nido-carboranyl azide and its “click” reactions. Molecules 2021, 26, 530.
- Druzina, A.A.; Zhidkova, O.B.; Dudarova, N.V.; Nekrasova, N.A.; Suponitsky, K.Y.; Timofeev, S.V.; Bregadze, V.I. Synthesis of zwitter-ionic conjugate of nido-carborane with cholesterol. Molecules 2021, 26, 6687.
- Yang, Z.; Zhao, W.; Liu, W.; Wei, X.; Chen, M.; Zhang, X.; Zhang, X.; Liang, Y.; Lu, C.; Yan, H. Metal-free oxidative B-N coupling of nido-carborane with N-heterocycles. Angew. Chem. Int. Ed. 2019, 58, 11886–11892.
- Yang, L.; Jei, B.B.; Scheremetjew, A.; Kuniyil, R.; Ackermann, L. Electrochemical B-H nitrogenation: Access to amino acids and BODIPY-labeled nido-carboranes. Angew. Chem. Int. Ed. 2021, 60, 1482–1487.
- Chen, M.; Zhao, D.; Xu, J.; Li, C.; Lu, C.; Yan, H. Electrooxidative B-H functionalization of nido-carboranes. Angew. Chem. Int. Ed. 2021, 60, 7838–7844.
- Huang, R.; Zhao, W.; Xu, S.; Xu, J.; Li, C.; Lu, C.; Yan, H. Photoredox B-H functionalization to selective B-N(sp3) coupling of nido-carborane with primary and secondary amines. Chem. Commun. 2021, 57, 8580–8583.
- Anufriev, S.A.; Sivaev, I.B.; Suponitsky, K.Y.; Godovikov, I.A.; Bregadze, V.I. Synthesis of 10-methylsulfide and 10-alkylmethylsulfonium nido-carborane derivatives: B–H π interactions between the B–H–B hydrogen atom and alkyne group in 10-RC≡CCH2S(Me)-7,8-C2B9H11. Eur. J. Inorg. Chem. 2017, 2017, 4436–4443.
- Erokhina, S.A.; Stogniy, M.Y.; Suponitsky, K.Y.; Kosenko, I.D.; Sivaev, I.B.; Bregadze, V.I. Synthesis of new nido-carborane based carboxylic acids and amines. Polyhedron 2018, 153, 145–151.
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Anisimov, A.A.; Sivaev, I.B.; Bregadze, V.I. Nucleophilic addition reactions to the ethylnitrilium derivative of nido-carborane 10-EtC≡N-7,8-C2B9H11. New J. Chem. 2018, 42, 17958–17967.
- Stogniy, M.Y.; Erokhina, S.A.; Anisimov, A.A.; Suponitsky, K.Y.; Sivaev, I.B.; Bregadze, V.I. 10-NCCH2CH2OCH2CH2C≡N-7,8-C2B9H11: Synthesis and reactions with various nucleophiles. Polyhedron 2019, 174, 114170.
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Anisimov, A.A.; Godovikov, I.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of novel carboranyl amidines. J. Organomet. Chem. 2020, 909, 121111.
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Markov, V.Y.; Sivaev, I.B. Synthesis and crystal structures of nickel(II) and palladium(II) complexes with o-carboranyl amidine ligands. Dalton Trans. 2021, 50, 4967–4975.
- Semioshkin, A.A.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of polyhedral boron hydrides and their synthetic applications. Dalton Trans. 2008, 977–992.
- Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives as an efficient synthetic tool for the modification of polyhedral boron hydrides. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 623–637.
- Druzina, A.A.; Shmalko, A.V.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of cobalt and iron bis(dicarbollide)s and their use in organic synthesis. Russ. Chem. Rev. 2021, 90, 785–830.
- Young, D.C.; Howe, D.V.; Hawthorne, M.F. Ligand derivatives of (3)-1,2-dicarbadodecahydroundecaborate(-1). J. Am. Chem. Soc. 1969, 91, 859–862.
- Stogniy, M.Y.; Abramova, E.N.; Lobanova, I.A.; Sivaev, I.B.; Bragin, V.I.; Petrovskii, P.V.; Tsupreva, V.N.; Sorokina, O.V.; Bregadze, V.I. Synthesis of functional derivatives of 7,8-dicarba-nido-undecaborate anion by ring-opening of its cyclic oxonium derivatives. Collect. Czech. Chem. Commun. 2007, 72, 1676–1688.
- Zakharkin, L.I.; Kalinin, V.N.; Zhigareva, G.G. Oxidation of dicarbadodecahydronidoundecaborate anions by mercury chloride in tetrahydrofuran and pyridine. Bull. Acad. Sci. USSR Div. Chem. Sci. 1979, 28, 2198–2199.
- Stogniy, M.Y.; Sivaev, I.B.; Malysheva, Y.B.; Bregadze, V.I. Synthesis of tetrahydropyran oxonium derivative of 7,8-dicarba-nido-undecaborane anion . Vestnik N. I. Lobachevskiy Nizhegorod Univ. 2013, 4, 115–117. Available online: http://www.unn.ru/pages/e-library/vestnik/99999999_West_2013_4(1)/19.pdf (accessed on 30 December 2021).
- Laskova, J.; Kosenko, I.; Serdyukov, A.; Sivaev, I.; Bregadze, V.I. Synthesis of naphthalimide derivatives of closo-dodecaborate and nido-carborane. J. Organomet. Chem. 2022, 959, 122186.
- Sivaev, I.B.; Stogniy, M.Y. Mercury derivatives of polyhedral boranes, carboranes, and metallacarboranes. Russ. Chem. Bull. 2019, 68, 217–253.
- Colquhoun, H.M.; Greenhough, T.J.; Wallbridge, M.G.H. Carbaborane derivatives of the late- and post-transition elements. Part 2. Dicarbaundecaboranyl compounds of copper(I), gold(I), and mercury(II); the crystal and molecular structure of 3-triphenylphosphine-3-mercura-1,2-dicarbadodecaborane(II), a pseudo-σ-bonded metallacarbaborane. J. Chem. Soc. Dalton Trans. 1979, 619–628.
- Teixidor, F.; Ayllon, J.A.; Viñas, C.; Kivekäs, R.; Sillanpää, R.; Casabo, J. Mercury coordination to Exo-dithio-7,8-dicarba-nido-undecaborate derivatives. J. Organomet. Chem. 1994, 483, 153–157.
- Shaw, K.F.; Reid, B.D.; Welch, A.J. Synthesis and characterisation of metal complexes of ether carbaboranes. Molecular structures of d6 ML3, d8 ML2 and d10 ML complexes of mono- and di-ether C2B9 carbaborane ligands, showing the progressive importance of secondary M…O bonding. J. Organomet. Chem. 1994, 482, 207–220.
- Řezácová, P.; Pokorná, J.; Brynda, J.; Kožíšek, M.; Cígler, P.; Lepšík, M.; Fanfrlík, J.; Řezáč, J.; Šašková, K.G.; Sieglová, I.; et al. Design of HIV protease inhibitors based on inorganic polyhedral metallacarboranes. J. Med. Chem. 2009, 52, 7132–7141.
- Frank, R.; Auer, H.; Hey-Hawkins, E. Functionalization of the nido-dicarbaborate anion nido-7,8-C2B9H12− by hydride abstraction. J. Organomet. Chem. 2013, 747, 217–224.
- Plešek, J.; Jelínek, T.; Mareš, F.; Heřmánek, S. Unique dialkylsulfoniomethylation of the 7,8-C2B9H12− ion to the 9-R2S-CH2-7,8-C2B9H11 zwitterions by formaldehyde and dialkyl sulfides. General synthesis of the compounds 10-R2E-7,8-C2B9H11 (E = O, S). Collect. Czech. Chem. Commun. 1993, 58, 1534–1547.
- Bakardjiev, M.; El Anwar, S.; Bavol, D.; Růžičková, Z.; Grüner, B. Focus on chemistry of the 10-dioxane-nido-7,8-dicarba-undecahydrido undecaborate zwitterion; exceptionally easy abstraction of hydrogen bridge and double-action pathways observed in ring cleavage reactions with OH− as nucleophile. Molecules 2020, 25, 814.
- Stogniy, M.Y.; Erokhina, S.A.; Kosenko, I.D.; Semioshkin, A.A.; Sivaev, I.B. Dimethyloxonium and methoxy derivatives of nido-carborane and metal complexes thereof. Inorganics 2019, 7, 46.
- Shmal’ko, A.V.; Anufriev, S.A.; Anisimov, A.A.; Stogniy, M.Y.; Sivaev, I.B.; Bregadze, V.I. Synthesis of cobalt and nickel 6,6′-diphenylbis(dicarbollides). Russ. Chem. Bull. 2019, 68, 1239–1247.
- Stogniy, M.Y.; Anufriev, S.A.; Bogdanova, E.V.; Sivaev, I.B.; Bregadze, V.I. Mercury(II) chloride in the synthesis of nido-carborane derivatives with B-N, B-O and B-S bonds. Russ. Chem. Bull. 2022, 71, 91–101.
- Stogniy, M.Y.; Anufriev, S.A.; Shmal’ko, A.V.; Antropov, S.M.; Anisimov, A.A.; Suponitsky, K.Y.; Filippov, O.A.; Sivaev, I.B. The unexpected reactivity of 9-iodo-nido-carborane: From nucleophilic substitution reactions to the synthesis of tricobalt tris(dicarbollide) Na. Dalton Trans. 2021, 50, 2671–2688.
More
Information
Subjects:
Chemistry, Inorganic & Nuclear
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
10 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No