Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandro Russo | + 2654 word(s) | 2654 | 2022-01-18 03:01:54 | | | |
| 2 | Yvaine Wei | Meta information modification | 2654 | 2022-01-29 02:41:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Russo, A. Hallmarks and Antimicrobic Therapy Management of Sepsis Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/18947 (accessed on 29 June 2026).
Russo A. Hallmarks and Antimicrobic Therapy Management of Sepsis Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/18947. Accessed June 29, 2026.
Russo, Alessandro. "Hallmarks and Antimicrobic Therapy Management of Sepsis Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/18947 (accessed June 29, 2026).
Russo, A. (2022, January 28). Hallmarks and Antimicrobic Therapy Management of Sepsis Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/18947
Russo, Alessandro. "Hallmarks and Antimicrobic Therapy Management of Sepsis Pathogenesis." Encyclopedia. Web. 28 January, 2022.
Copy Citation
Sepsis is a life-threatening condition that arises when the body’s response to an infection injures its own tissues and organs. Despite significant morbidity and mortality throughout the world, its pathogenesis and mechanisms are not clearly understood.
sepsis
pathogenesis
management
diagnosis
antibiotic therapy
1. Introduction
Sepsis is an important syndrome associated with significant morbidity and mortality. The true extent of sepsis is not fully understood due to its variability and the lack of specific epidemiological data, due to different diagnostic criteria and definitions. The World Health Organization (WHO) has stated that the worldwide annual mortality due to sepsis is around 6 million, with most of these deaths being preventable [1][2][3][4][5][6][7]. Originally defined as “the decomposition of animal or vegetable matter in the presence of bacteria” [8], “sepsis” is currently identified as “a life-threatening condition that arises when the body’s response to an infection injures its own tissues and organs” [9]. Initiated by an invading pathogen, generally represented by bacteria and, less frequently, by viruses or fungi, sepsis results in an inflammatory process in which the body’s own response has a deleterious effect upon itself. This pathophysiological response can culminate in multiorgan failure, usually due to a combination of cardiovascular, cellular, coagulation and endothelial dysfunction [10], eventually leading to septic shock, a clinical proinflammatory response, predominantly cytokine-mediated (see Figure 1).
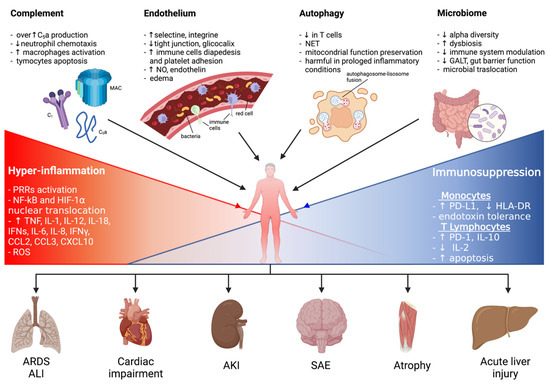
Figure 1. A graphical summary of the pathophysiological mechanisms involved in sepsis onset and persistence and related organ injuries. ALI: acute lung injury; AKI: acute kidney injury; ARDS: acute respiratory distress syndrome; CCL: C-C motif chemokine ligand; CXCL: C-X-C motif chemokine ligand; ET: endotoxin tolerance; GALT: gut-associated lymphoid tissue; HIF-1α: hypoxia-inducible factor-1α; HLA-DR: human leukocyte antigen-DR isotype; IFNγ: interferon γ; IL: interleukin; LPS: lipopolysaccharide; MAC: membrane attack complex; NET: neutrophil extracellular trap; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NO: nitric oxide; PD-1: programmed cell death 1 receptor; PD-L1: programmed cell death ligand 1; PRRs: pattern recognition receptors; ROS: reactive oxygen species; SAE: sepsis-associated encephalopathy.
2. Pathophysiology of Sepsis
2.1. The Innate Immunity
The mechanisms resulting in the development of sepsis are very complex and not completely understood. However, it is well established that at the beginning of sepsis the inflammatory response is mediated by the activation of the innate immune system cells, mainly represented by macrophages, monocytes, neutrophils, and natural killer cells. Multiple infection-derived microbial products are simultaneously recognized by complement and specific cell-surface receptors. Amongst these, toll-like receptors (TLRs) are transmembrane receptors expressed by monocytes and macrophages and able to detect extracellular pathogen-associated molecular patterns (PAMPs) (such as bacterial endotoxins and fungal β-glucans) and damage-associated molecular patterns (DAMPs) released from injured endogenous cells (such as ATP, high mobility group proteins, and mitochondrial DNA). To this extent, nod-like receptors (NODs), which are expressed intracellularly, recognize pathogens invading the cytosol. Furthermore, retinoic acid inducible gene (RIG)-like receptors, mannose-binding lectin (MBL), and scavenger receptors also take part in this process [11].
Therefore, the binding between cellular receptors and different components of bacteria, viruses, and fungi, as well as host products derived from tissue damage, induces multiple intracellular signaling pathways, ultimately leading to the expression of several common gene classes involved in inflammation, adaptive immunity, and cellular metabolism, the second key step in the activation of the immune response during sepsis.
2.2. The Complement System
The complement system, which consists of multiple proteins in body fluids, receptors, and regulatory proteins, carries out a defensive action against infectious agents and acts as an immune sensor, effector, and regulator. Complement activation can be initiated via three different pathways: the classical (including antibodies, C1q, C2, and C4), the alternative (including complement factor B and spontaneous C3 hydrolysis to form C3b), and the lectin pathway (including MBL and ficolins) [12][13]. The common result of these pathways is the cleavage of C3 and C5 to generate anaphylatoxin peptides (i.e., C3a and C5a), C3b, and C5b. C3b is an important phagocytosis-promoting product, whereas C5b interacts with C6–C9 to form the membrane attack complex on cell membranes. C5a, under conditions of regulated production, supplies defensive functions by enhancing chemotactic responses of neutrophils, phagocytosis, and oxidative burst that is involved in killing bacteria [14][15][16].
The role of complement in sepsis pathogenesis might appear ambiguous. On one hand, C3 deficiency, which results in the inhibition of most complement effector functions, clearly increases sepsis-associated mortality in animals [17]; these observations underline the pivotal role of complement as a defense mechanism against invading microbes. Contrariwise, other data have indicated that inhibition of C5a signaling improves the survival of experimental animal models [18][19]. Increased production of C5a, as occurs during sepsis, can lead to adverse systemic consequences. Neutrophils become functionally paralyzed [20], unable to respond chemotactically to C5a, but also to the chemotactic peptide N-formyl-Met-Leu-Phe (fMLP), which is produced by bacteria [21].
2.3. The Role of the Endothelium
Sepsis is not only a state of systemic inflammation, but also a state of deregulated hemostasis. Hemostasis is a complex process regulated by the endothelium, soluble plasma molecules, platelets, and leukocytes; it not only is involved in the balance between pro- and anticoagulant forces, but also directs platelet and fibrin clotting to areas of focal vascular injury [22]. Sustained inflammation during severe sepsis drives hemostasis in a condition of deregulation characterized by a prothrombotic and antifibrinolytic state, organ ischemia, and multiple organ dysfunction syndrome.
Sepsis is associated with severe endothelium dysfunction leading to deregulation of vascular reactivity and hemostasis. This damage of the endothelial cells (ECs) is considered pivotal to the progression to organ failure during sepsis. Under normal conditions, the endothelium serves as an anticoagulant surface that regulates the flow of gases, water, solutes, lipids, proteins, and other macromolecules within the microcirculation [23][24]. The endothelium integrity is maintained by the cell cytoskeleton (actin), intercellular adhesion molecules (tight junctions), and numerous supportive proteins. During sepsis, these structures are broken up essentially in response to neutrophil and platelet adhesion, the release of inflammatory mediators, and toxic intermediates.
2.4. Autophagy
Autophagy is a highly conserved degradative pathway involved in maintaining intracellular homeostasis under physiological conditions, playing a crucial role in the pathogenesis of inflammation and infectious diseases [25][26]. There are three types of autophagy: macroautophagy, chaperone-mediated autophagy, and microautophagy. To eliminate damaged proteins and organelles, as well as cytoplasmatic bacteria and pathogens [27], cells exploit this adaptive mechanism to protect themselves from damages and apoptosis [28]. Several intracellular signaling pathways are responsible for autophagy induction, such as 5′ adenosine monophosphate-activated protein kinase (AMPK) and c-Jun N-terminal kinase (JNK)/p38, two MAPK pathways, generating ROS and regulating the NF-κB under activation of TLR4 and TLR9, respectively [29][30]. Recently, the induction of autophagy has received increased attention in the context of sepsis: it mainly protects the host against multiorgan dysfunction syndrome (MODS) by preventing immune cell apoptosis, maintaining the homeostatic balance between pro- and anti-inflammatory cytokines, and preserving mitochondrial functions [31][32][33][34].
2.5. Downregulation of the Immune System
Besides the systemic inflammatory response characterizing sepsis during the early stages of the process, a prolonged state of immunosuppression also occurs in both the initial and late phases of the disease [35][36]. Indeed, patients who survive the early inflammatory stage of sepsis enter a late phase characterized by profound immunosuppression: these patients frequently experience ongoing infectious foci, despite antimicrobial therapy; reactivation of latent viral infection; and acquisition of secondary hospital-acquired infections, often with opportunistic microorganisms, which usually do not tend to infect patients with normal immune status.
Sepsis has been described as a two-phase process where an initial hyperinflammatory phase is followed by a prolonged immunosuppressive phase [37][38]. However, in clinical practice, it is evident that these two phases tend to overlap. Indeed, several studies have shown that both proinflammatory and anti-inflammatory responses occur simultaneously in the first stage of sepsis [39][40][41]. The net effect of such mechanisms results in immunosuppression involving both the innate and the adaptive immune systems.
2.6. The Role of the Microbiome
The microbiome is the microbic consortium of bacteria, viruses, fungi, and protozoa living upon (skin) and inside (gut, lung) our body. The last few decades have seen an increasing interest regarding the physiological and pathogenetic role of the microbiome in several medical conditions, included sepsis. Indeed, the gut microbiome exerts numerous physiological roles as an independent organ within the human body: it produces functionally active metabolites which influence immune functions of several immune cells; it increases the gut barrier function, inhibiting hematic translocations of resident microorganisms; and it directly promotes the maturation of local immune cells, driving the expansion of antigen-specific activated T cells and enhancing responsiveness of immune cells to cytokines [42].
2.7. Cellular, Tissue, and Organ Failure
Sepsis is also described as a systemic disorder, affecting all organs of the body. Although the molecular basis of organ failure remains unclear, six types of organ dysfunction predominantly characterize sepsis: neurological (altered mental status), pulmonary (hypoxemia), cardiovascular (shock), renal (oliguria and/or increased creatinine concentration), hematological (decreased platelet count), and hepatic (hyperbilirubinemia). The underlying mechanism behind tissue and organ dysfunction in sepsis seems to be a diminished oxygen delivery to and utilization by cells with a consequent increased anaerobic glycolysis and lactic acid production. Several factors, including hypotension, reduced red-cell deformability, and microvascular thrombosis, contribute to impair tissue oxygenation in septic shock in addition to mitochondrial damage caused by oxidative stress [43]. All these mechanisms in conjunction with systemic hyperinflammation and sustained immunosuppression, generalized increased catabolism, insulin resistance, and hyperglycemia can contribute to the cellular level damage.
3. Diagnosis of Sepsis
3.1. MDR and Sepsis
In the last few decades, a specific sepsis population with a high mortality risk is accounted for by patients with septic shock by multidrug-resistant (MDR) microorganisms, with Gram-negative pathogens being responsible for most cases [44]. In particular, an increased frequency of MDR Gram-negative pathogens, such as MDR Acinetobacter baumannii (MDR-AB) and Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae (KPC-Kp), have been observed among critically ill intensive care unit (ICU) patients.
Although several risk factors for MDR microorganism infections have been identified (see Table 1), the real causes for this increased risk are still unclear, and the appropriateness of initial antibiotic therapy still represents a crucial variable in septic patients, thus affecting the clinical outcome [45][46][47][48].
Table 1. Main risk factors for MDR infections.
| Advanced age |
| Diabetes |
| End-stage liver disease |
| Immunosuppressive therapy |
| Use of corticosteroids |
| Malignancy |
| Organ transplantation |
| Recent surgery |
| Recent exposure (<3 months) to antibiotic therapy |
| Prior hospital admission |
| MDR colonization |
| Local epidemiology |
Legend. MDR: multidrug-resistant.
3.2. Diagnostic Tools
The ideal diagnostic technology for sepsis should include the following characteristics: a rapid and broad-based detection, minimal invasiveness, clinical sample usage with low specimen volumes, high sensitivity and specificity for the immediate initiation of targeted antibiotic use in the presence of signs and symptoms of systemic inflammation, and detection of drug resistance and unknown and emerging pathogens. So far, blood culture is commonly known as the gold standard for the detection of microbial pathogens in the bloodstream. However, the organisms’ growth to detectable levels in routine blood cultures can take up to 5 days, with additional time being required for identification (24 h) and testing for antibiotic susceptibility (48 h) [49][50][51][52]. False positives via contamination during sample collection (e.g., Staphylococcus epidermidis) are also common. Moreover, adults with bacteremia and/or with fungemia might receive an inappropriate treatment before microbiology culture results become available [53].
4. Therapeutic Approach
4.1. General Considerations
Besides supportive therapy (vasopressor administration, mechanical ventilation, renal replacement therapy), the treatment of sepsis and septic shock is based on empirical antibiotic therapy and infection source control. The 2021 Surviving Sepsis Campaign provides recommendations about the management of sepsis and septic shock [54]. As reported above, it is crucial to collect blood cultures prior to antibiotic administration and to start therapy with broad-spectrum antibiotics within 1 h after recognition of sepsis or septic shock condition. In 2014, the MEDUSA trial showed that delay in antimicrobial therapy and source control was associated with increased mortality in sepsis and septic shock patients: every hour the antibiotic therapy was delayed, mortality increased by 2% [55][56].
Nevertheless, it is important to administer appropriate antibiotic therapy rather than early administration of any antibiotic.
4.2. Patients in ICU
Critically ill patients with severe sepsis present a significant fluid shift from the intravascular compartment to interstitial space, caused by aggressive fluid resuscitation and hyperdynamic state associated with sepsis itself [57][58][59][60][61][62][63][64][65][66][67][68][69]. Extracellular fluid changes may also be enhanced by edematous states, pleural effusion, postsurgical drains, and extracorporeal membrane oxygenation.
Time-dependent antibiotics, which reach maximal efficacy when their concentration exceeds the MIC for a longer time, are mostly hydrophilic, so they result underdosed when Vd and drug clearance are increased. Such is the case of β-lactams, whose antimicrobial activity is optimized by more frequent administration rather than higher doses. Studies in ICUs have demonstrated that extended (3–4 h) or continuous (24 h) infusions of β-lactam antibiotics have equivalent or improved outcomes compared to intermittent (0.5–1 h) infusions, without increased adverse events.
4.3. Patients with Altered Renal Clearance
Dose adjustment is routinely recommended in patients with impaired renal function. Nevertheless, critically ill patients often present an augmented renal clearance (ARC) with a 130–160 mL/min creatinine clearance, due to aggressive fluid resuscitation, increased cardiac output, vasopressor use, and enhanced kidney blood flow. Patients with ARC are more likely to be younger (age < 50 years), male, have a modified Sequential Organ Failure Assessment Score (SOFA < 4 or less), and be admitted because of trauma [69]. This condition requires antimicrobial dose adjustments to avoid subtherapeutic concentrations, especially for β-lactams. Unfortunately, dose increase in these patients is not standard practice.
Another condition that may profoundly affect antibiotic dosing is continuous renal replacement therapy (CRRT), used for AKI management in hemodynamically unstable critically ill patients. Indeed, severe sepsis and septic shock are among the two most common reasons for CRRT initiation. CRRT can differ in modalities, hemofilters, and effluent rates, all of which may require dosing adjustments [70].
4.4. Obese Patients
Clinical studies report conflicting results about the impact of obesity on mortality in critically ill sepsis patients. Obesity is known as a chronic inflammation state that is related to increased oxidative stress [71]. A multitude of physiologic changes affecting PK and PD can occur in obese patients and may be responsible for antibiotic treatment failure due to lower serum concentration.
4.5. Burn Patients
Burn patients represent a particular population of critically ill patients. They are more susceptible to acquiring infections, and sepsis is the most important cause of mortality (rates of sepsis-related death are 50–84% in adult burn patients) [72]. This increased susceptibility has been attributed to some causes such as a nonspecific immunosuppressive state induced by burns (myeloid maturation arrest causing neutropenia, compromised cytotoxic T lymphocyte response, impaired neutrophil function, and decreased macrophage production [73]), loss of skin protection, respiratory injury from smoke, and frequent use of invasive devices (tracheal intubation, intravascular and urinary catheters) [74]. The leading cause of sepsis in these patients is the infection of burn wounds, and the most common isolated organisms are Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus [72].
4.6. Adjunctive Therapies
Since sepsis and septic shock are characterized by a dysfunction of the immune response, with an initial increase in proinflammatory cytokines and a subsequent immune-paralysis, adjunctive immune-modulatory treatments have been developed in support of antibiotic therapies to restore immune response. Single adjunctive therapies have been studied and are currently being evaluated in clinical trials, with discordant results. Nevertheless, an observational study by Marik et al. showed a synergistic effect of a combination of intravenous vitamin C, thiamine, and hydrocortisone, resulting in a reduction in organ dysfunction and mortality of patients with septic shock [75].
5. Take-Home Messages
In Figure 2 are reported most important take home messages from this entry.
Figure 2. “Take home messages” about the main points treated in this entry.
-
The mechanisms of sepsis are mainly based on the activation of a hyperinflammatory innate immune system response to infective stimuli and consequent endothelial activation and humoral changes, but it mostly relies on immunosuppression mechanisms involving both the innate and the adaptive immune systems.
-
The gold-standard diagnostic laboratory technique for the diagnosis of sepsis remains blood cultures.
-
Procalcitonin is an important tool to differentiate sepsis from noninfectious diseases and thereby contribute to early diagnosis.
-
Prompt empirical broad-spectrum antibiotic therapy and source control of infection are the most effective treatment strategy in sepsis.
-
Pharmacokinetic/pharmacodynamic adjustments are recommended for patients with specific characteristics (obesity, burns, altered renal function).
References
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing Sepsis as a Global Health Priority—A WHO Resolution. N. Engl. J. Med. 2017, 377, 414–417.
- World Health Organization. Improving the Prevention, Diagnosis and Clinical Management of Sepsis. 2017. Available online: http://apps.who.int/gb/ebwha/pdf_files/WHA70/A70_R7-en.pdf (accessed on 15 November 2021).
- Weinstein, M.P.; Reller, L.B.; Murphy, J.R.; Lichtenstein, K.A. The Clinical Significance of Positive Blood Cultures: A Comprehensive Analysis of 500 Episodes of Bacteremia and Fungemia in Adults. I. Laboratory and Epidemiologic Observations. Clin. Infect. Dis. 1983, 5, 35–53.
- Lee, C.-C.; Chen, S.-Y.; Chang, I.-J.; Chen, S.-C.; Wu, S.-C. Comparison of Clinical Manifestations and Outcome of Community-Acquired Bloodstream Infections Among the Oldest Old, Elderly, and Adult Patients. Medicine 2007, 86, 138–144.
- Weinstein, M.P.; Towns, M.L.; Quartey, S.M.; Mirrett, S.; Reimer, L.G.; Parmigiani, G.; Reller, L.B. The Clinical Significance of Positive Blood Cultures in the 1990s: A Prospective Comprehensive Evaluation of the Microbiology, Epidemiology, and Outcome of Bacteremia and Fungemia in Adults. Clin. Infect. Dis. 1997, 24, 584–602.
- Elixhauser, A.; Friedman, B.; Stranges, E. Septicemia in U.S. Hospitals, 2009: Statistical Brief #122. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2011.
- Buehler, S.S.; Madison, B.; Snyder, S.R.; Derzon, J.; Cornish, N.E.; Saubolle, M.A.; Weissfeld, A.S.; Weinstein, M.P.; Liebow, E.B.; Wolk, D.M. Effectiveness of Practices to Increase Timeliness of Providing Targeted Therapy for Inpatients with Bloodstream Infections: A Laboratory Medicine Best Practices Systematic Review and Meta-analysis. Clin. Microbiol. Rev. 2016, 29, 59–103.
- Geroulanos, S.; Douka, E.T. Historical perspective of the word “sepsis”. Intensiv. Care Med. 2006, 32, 2077.
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810.
- Evans, T. Diagnosis and management of sepsis. Clin. Med. 2018, 18, 146–149.
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820.
- Carroll, M.C.; Isenman, D.E. Regulation of Humoral Immunity by Complement. Immunity 2012, 37, 199–207.
- Sacks, S.H.; Zhou, W. The role of complement in the early immune response to transplantation. Nat. Rev. Immunol. 2012, 12, 431–442.
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Genet. 2008, 6, 132–142.
- Markiewski, M.M.; Lambris, J.D. The Role of Complement in Inflammatory Diseases from Behind the Scenes into the Spotlight. Am. J. Pathol. 2007, 171, 715–727.
- Guo, R.-F.; Ward, P.A. Role of C5A in inflammatory responses. Annu. Rev. Immunol. 2005, 23, 821–852.
- Wessels, M.R.; Butko, P.; Ma, M.; Warren, H.B.; Lage, A.L.; Carroll, M.C. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA 1995, 92, 11490–11494.
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Day, D.E.; Zetoune, F.S.; Sarma, J.V.; Huber-Lang, M.S.; Ward, P.A. Functions of the complement components C3 and C5 during sepsis. FASEB J. 2008, 22, 3483–3490.
- Ward, P.A. Role of the complement in experimental sepsis. J. Leukoc. Biol. 2008, 83, 467–470.
- Huber-Lang, M.S.; Younkin, E.M.; Sarma, J.V.; McGuire, S.R.; Lu, K.T.; Guo, R.F.; Padgaonkar, V.A.; Curnutte, J.T.; Erickson, R.; Ward, P.A. Complement-Induced Impairment of Innate Immunity During Sepsis. J. Immunol. 2002, 169, 3223–3231.
- Tomhave, E.D.; Richardson, R.M.; Didsbury, J.R.; Menard, L.; Snyderman, R.; Ali, H. Cross-desensitization of receptors for peptide chemoattractants. Characterization of a new form of leukocyte regulation. J. Immunol. 1994, 153, 8089498.
- Deutschman, C.S.; Tracey, K.J. Sepsis: Current Dogma and New Perspectives. Immunity 2014, 40, 463–475.
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370.
- Schouten, M.; Wiersinga, W.J.; Levi, M.; Van Der Poll, T. Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol. 2007, 83, 536–545.
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101.
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354.
- Oami, T.; Watanabe, E.; Hatano, M.; Teratake, Y.; Fujimura, L.; Sakamoto, A.; Ito, C.; Toshimori, K.; Swanson, P.E.; Oda, S. Blocking Liver Autophagy Accelerates Apoptosis and Mitochondrial Injury in Hepatocytes and Reduces Time to Mortality in a Murine Sepsis Model. Shock 2018, 50, 427–434.
- Karagiannidis, I.; Kataki, A.; Glustianou, G.; Memos, N.; Papalois, A.; Alexakis, N.; Zografos, G.C.; Konstadoulakis, M.M. Extended Cytoprotective Effect of Autophagy in the Late Stages of Sepsis and Fluctuations in Signal Transduction Pathways in a Rat Experimental Model of Kidney Injury. Shock. 2016, 45, 139–147.
- Kim, M.J.; Kim, E.H.; Pun, N.T.; Chang, J.-H.; Kim, J.-A.; Jeong, J.-H.; Choi, D.Y.; Kim, S.-H.; Park, P.-H. Globular Adiponectin Inhibits Lipopolysaccharide-Primed Inflammasomes Activation in Macrophages via Autophagy Induction: The Critical Role of AMPK Signaling. Int. J. Mol. Sci. 2017, 18, 1275.
- Wu, H.-M.; Wang, J.; Zhang, B.; Fang, L.; Xu, K.; Liu, R.-Y. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci. 2016, 161, 51–59.
- Piquereau, J.; Godin, R.; Deschênes, S.; Bessi, V.L.; Mofarrahi, M.; Na Hussain, S.; Burelle, Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013, 9, 1837–1851.
- Yen, Y.-T.; Yang, H.-R.; Lo, H.-C.; Hsieh, Y.-C.; Tsai, S.-C.; Hong, C.-W.; Hsieh, C.-H. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery 2013, 153, 689–698.
- Ho, J.; Yu, J.; Wong, S.H.; Zhang, L.; Liu, X.; Wong, W.T.; Leung, C.; Choi, G.; Wang, M.H.; Gin, T.; et al. Autophagy in sepsis: Degradation into exhaustion? Autophagy 2016, 12, 1073–1082.
- Takahashi, W.; Watanabe, E.; Fujimura, L.; Watanabe-Takano, H.; Yoshidome, H.; Swanson, P.E.; Tokuhisa, T.; Oda, S.; Hatano, M. Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit. Care 2013, 17, R160.
- Munoz, C.; Carlet, J.; Fitting, C.; Misset, B.; Blériot, J.P.; Cavaillon, J.M. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Investig. 1991, 88, 1747–1754.
- Delano, M.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 2007, 204, 1463–1474.
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497.
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874.
- Munfod, R.S.; Pugin, J. Normal Responses to Injury Prevent Systemic Inflammation and Can Be Immunosuppressive. Am. J. Respir. Crit. Care Med. 2001, 163, 316–321.
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A genomic storm in critically injured humans. J. Exp. Med. 2011, 208, 2581–2590.
- Stearns-Kurosawa, D.J.; Osuchowski, M.F.; Valentine, C.; Kurosawa, S.; Remick, D.G. The Pathogenesis of Sepsis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 19–48.
- Kullberg, R.F.; Wiersinga, W.J.; Haak, B.W. Gut microbiota and sepsis: From pathogenesis to novel treatments. Curr. Opin. Gastroenterol. 2021, 37, 578–585.
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64.
- Cornaglia, G.; Giamarellou, H.; Rossolini, G.M. Metallo-β-lactamases: A last frontier for β-lactams? Lancet Infect. Dis. 2011, 11, 381–393.
- Akella, K.; Joshi, G.; Ibrahim, S.; Rutka, P.; Chow, P.; Fernando, R.; Sklarek, H. #1274: Degree of multidrug resistance in sepsis is associated with increased in-hospital morbidity. Crit. Care Med. 2019, 47, 613.
- Knaus, W.A.; Draper, E.A.; Wagner, D.P.; Zimmerman, J.E. APACHE II: A severity of disease classification system. Crit. Care Med. 1985, 13, 818–829.
- Karamouzos, V.; Giamarellos-Bourboulis, E.J.; Velissaris, D.; Gkavogianni, T.; Gogos, C. Cytokine production and outcome in MDR versus non-MDR gram-negative bacteraemia and sepsis. Infect. Dis. 2021, 53, 764–771.
- Lee, S.J.; You, J.S.; Gharbi, A.; Kim, Y.J.; Lee, M.S.; Kim, D.H.; Lee, K.W.; Jung, I.D.; Park, Y.M. IOX1 activity as sepsis therapy and an antibiotic against multidrug-resistant bacteria. Sci. Rep. 2021, 11, 1–12.
- Riedel, S.; Carroll, K.C. Early Identification and Treatment of Pathogens in Sepsis: Molecular Diagnostics and Antibiotic Choice. Clin. Chest Med. 2016, 37, 191–207.
- Bacconi, A.; Richmond, G.S.; Baroldi, M.A.; Laffler, T.G.; Blyn, L.B.; Carolan, H.E.; Frinder, M.R.; Toleno, D.M.; Metzgar, D.; Gutierrez, J.R.; et al. Improved Sensitivity for Molecular Detection of Bacterial and Candida Infections in Blood. J. Clin. Microbiol. 2014, 52, 3164–3174.
- Opota, O.; Jaton, K.; Greub, G. Microbial diagnosis of bloodstream infection: Towards molecular diagnosis directly from blood. Clin. Microbiol. Infect. 2015, 21, 323–331.
- Connell, T.G.; Rele, M.; Cowley, D.; Buttery, J.P.; Curtis, N. How Reliable Is a Negative Blood Culture Result? Volume of Blood Submitted for Culture in Routine Practice in a Children’s Hospital. Pediatrics 2007, 119, 891–896.
- Sinha, M.; Jupe, J.; Mack, H.; Coleman, T.P.; Lawrence, S.M.; Fraley, S.I. Emerging Technologies for Molecular Diagnosis of Sepsis. Clin. Microbiol. Rev. 2018, 31, e00089-17.
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock. Crit. Care Med. 2021, 49, e1063–e1143.
- Jarczak, D.; Kluge, S.; Nierhaus, A. Sepsis—Pathophysiology and Therapeutic Concepts. Front. Med. 2021, 8, 8302.
- Richter, D.C.; Heininger, A.; Brenner, T.; Hochreiter, M.; Bernhard, M.; Briegel, J.; Dubler, S.; Grabein, B.; Hecker, A.; Krüger, W.A.; et al. Bacterial sepsis: Diagnostics and calculated antibiotic therapy. Der Anaesthesist 2017, 66, 737–761.
- Buckman, S.A.; Turnbull, I.R.; Mazuski, J.E. Empiric Antibiotics for Sepsis. Surg. Infect. 2018, 19, 147–154.
- Esposito, S.; De Simone, G.; Boccia, G.; De Caro, F.; Pagliano, P. Sepsis and septic shock: New definitions, new diagnostic and therapeutic approaches. J. Glob. Antimicrob. Resist. 2017, 10, 204–212.
- Sjövall, F.; Perner, A.; Møller, M.H. Empirical mono- versus combination antibiotic therapy in adult intensive care patients with severe sepsis—A systematic review with meta-analysis and trial sequential analysis. J. Infect. 2017, 74, 331–344.
- Russo, A.; Bassetti, M.; Bellelli, V.; Bianchi, L.; Cattaneo, F.M.; Mazzocchetti, S.; Paciacconi, E.; Cottini, F.; Schiattarella, A.; Tufaro, G.; et al. Efficacy of a Fosfomycin-Containing Regimen for Treatment of Severe Pneumonia Caused by Multidrug-Resistant Acinetobacter baumannii: A Prospective, Observational Study. Infect. Dis. Ther. 2021, 10, 187–200.
- Dugar, S.; Choudhary, C.; Duggal, A. Sepsis and septic shock: Guideline-based management. Clevel. Clin. J. Med. 2020, 87, 53–64.
- Coopersmith, C.M.; De Backer, D.; Deutschman, C.S.; Ferrer, R.; Lat, I.; Machado, F.R.; Martin, G.S.; Martin-Loeches, I.; Nunnally, M.E.; Antonelli, M. Surviving sepsis campaign: Research priorities for sepsis and septic shock. Intensiv. Care Med. 2018, 44, 1400–1426.
- Allison, M.G.; Heil, E.L.; Hayes, B.D. Appropriate Antibiotic Therapy. Emerg. Med. Clin. N. Am. 2017, 35, 25–42.
- Williams, J.M.; Keijzers, G.; Macdonald, S.P.; Shetty, A.; Fraser, J.F. Review article: Sepsis in the emergency department—Part 3: Treatment. Emerg. Med. Australas. 2018, 30, 144–151.
- Veiga, R.P.; Paiva, J.-A. Pharmacokinetics–pharmacodynamics issues relevant for the clinical use of beta-lactam antibiotics in critically ill patients. Crit. Care 2018, 22, 1–34.
- Ahmed, N.; Jen, S.-P.; Altshuler, D.; Papadopoulos, J.; Pham, V.; Dubrovskaya, Y. Evaluation of Meropenem Extended Versus Intermittent Infusion Dosing Protocol in Critically Ill Patients. J. Intensiv. Care Med. 2018, 35, 763–771.
- Burger, R.; Guidi, M.; Calpini, V.; Lamoth, F.; Decosterd, L.; Robatel, C.; Buclin, T.; Csajka, C.; Marchetti, O. Effect of renal clearance and continuous renal replacement therapy on appropriateness of recommended meropenem dosing regimens in critically ill patients with susceptible life-threatening infections. J. Antimicrob. Chemother. 2018, 73, 3413–3422.
- Nielsen, E.I.; Cars, O.; Friberg, L. Pharmacokinetic/Pharmacodynamic (PK/PD) Indices of Antibiotics Predicted by a Semimechanistic PKPD Model: A Step toward Model-Based Dose Optimization. Antimicrob. Agents Chemother. 2011, 55, 4619–4630.
- Heffernan, A.; Sime, F.; Taccone, F.S.; Roberts, J.A. How to optimize antibiotic pharmacokinetic/pharmacodynamics for Gram-negative infections in critically ill patients. Curr. Opin. Infect. Dis. 2018, 31, 555–565.
- Shaw, A.R.; Chaijamorn, W.; Mueller, B.A. We Underdose Antibiotics in Patients on CRRT. Semin. Dial. 2016, 29, 278–280.
- Trivedi, V.; Bavishi, C.; Jean, R. Impact of obesity on sepsis mortality: A systematic review. J. Crit. Care 2015, 30, 518–524.
- Lopez, O.N.; Cambiaso-Daniel, J.; Branski, L.K.; Norbury, W.B.; Herndon, D.N. Predicting and managing sepsis in burn patients: Current perspectives. Ther. Clin. Risk Manag. 2017, 13, 1107–1117.
- Avni, T.; Levcovich, A.; Ad-El, D.D.; Leibovici, L.; Paul, M. Prophylactic antibiotics for burns patients: Systematic review and meta-analysis. BMJ 2010, 340, c241.
- Hidalgo, F.; Mas, D.; Rubio, M.; Garcia-Hierro, P. Infections in critically ill burn patients. Med. Intensiv. 2016, 40, 179–185.
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, Vitamin C, and Thiamine for the Treatment of Severe Sepsis and Septic Shock. Chest 2017, 151, 1229–1238.
More
Information
Subjects:
Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
564
Revisions:
2 times
(View History)
Update Date:
06 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No