Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vic Norris | + 3944 word(s) | 3944 | 2022-01-25 03:18:56 | | | |
| 2 | Bruce Ren | Meta information modification | 3944 | 2022-01-28 02:00:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Norris, V. A Defective Viral Particle Approach to COVID-19. Encyclopedia. Available online: https://encyclopedia.pub/entry/18902 (accessed on 19 June 2026).
Norris V. A Defective Viral Particle Approach to COVID-19. Encyclopedia. Available at: https://encyclopedia.pub/entry/18902. Accessed June 19, 2026.
Norris, Vic. "A Defective Viral Particle Approach to COVID-19" Encyclopedia, https://encyclopedia.pub/entry/18902 (accessed June 19, 2026).
Norris, V. (2022, January 27). A Defective Viral Particle Approach to COVID-19. In Encyclopedia. https://encyclopedia.pub/entry/18902
Norris, Vic. "A Defective Viral Particle Approach to COVID-19." Encyclopedia. Web. 27 January, 2022.
Copy Citation
Defective interfering particles, which arise naturally and interfere with viruses, have long inspired the idea that they might be adapted to treat viral diseases. Here, we explore how such defective interfering particles and other therapeutic nanoparticles might be designed and constructed to interfere with SARS-CoV-2.
antivirus
immunity
therapy
coronavirus
defective interfering particle
aptamer
COVID-19
extracellular vesicle
synthetic defective viral genome
1. Introduction
In a Holliday lecture in 1999, Don Ganem explained that pandemic infection is a recurrent, not a temporary, phenomenon: “The supervention of the AIDS pandemic put the lie to all of these optimistic predictions about how infectious disease was conquered and was no longer a problem. Now we know, of course, that notion was foolish to begin with, that infectious disease, epidemic infection, is a part of the human condition. I’m going to show you that it’s really a part of human evolution that we can never get away from infectious disease as a class. We can triumph over individual infectious diseases, but the concept that we’re going to be free of infection as a species is a ridiculous one and one that nobody believes anymore” [1]. The current COVID-19 pandemic is a reminder that the need to develop new weapons to combat infections is as pressing now as then.
A powerful, anti-viral strategy could exploit the DIPs and related particles that arise naturally for every family virus. DIPs are particles containing degenerate forms of the virus genome, which interfere with the replication of the parental virus but are non-replicative per se. Historically, such particles were considered artefacts of virus propagation in vitro; however, studies have shown that defective viral genomes are present in patients infected with viruses such as hepatitis C, influenza A and respiratory syncytia [2][3][4]. Thus, DIPs are currently investigated for their potential role in influencing disease outcomes and shaping virus evolution. A similar form of interference is observed when small viruses such as satellite viruses and virophages parasitize the larger viruses with which they are associated thereby decreasing their fitness. Other particles that can be produced during an infection include the virus-like particles (VLPs). These particles are composed of viral structural proteins, they morphologically resemble the parental virus but are non-infectious due to lack of genetic material. Another type is the extracellular vesicle (EV). EVs are released by all types of cells as they facilitate intercellular communication. During an infection they communicate virus-specific signals.
DIPs reduce virulence, induce high levels of interferons, and promote viral persistence by mechanisms that are not well understood [5]. In the case of RNA viruses, the shorter genome of the particle may allow it to out-compete the wild-type virus [6]. Alternatively, specific changes to the particle’s genome confer advantages over the full-length genome in using a limiting viral or host factor [7]. Also, the particle’s genome may interfere with the assembly of the wild-type’s genome into virions [8]. Furthermore, the particles may stimulate the host immune system [9]. Finally, the particles may compete with the wild-type virus for entry or cause internalization of the virus entry receptor inhibiting virus entry and spread [5].
Therapeutic interfering particles (TIPs) are a class of DIPs engineered to reduce the severity of viral diseases [10]. TIPs could be designed to use the same transmission routes as the wild-type virus thereby limiting viral transmission in populations at risk. Given that DIPs are diverse, it is unclear which design of TIP would prove most effective. Single-cell studies have revealed the importance of phenotypic diversity in a large number of systems. It is likely that this also applies to viral infections; hence, the diverse population of DIPs actually benefits both the virus and host. A novel anti-viral strategy might take this into account by constructing a diverse population of synthetic defective viral genomes (synDVGs) with prophylactic or therapeutic potential. In addition, the properties of EVs and VLPs could be harnessed to suppress SARS-CoV-2 infection and mitigate pathogenesis. These particles could be engineered to trigger antiviral responses, alleviate inflammation, enable tissue regeneration, or interfere with macromolecular assemblies during SARS-CoV-2 infection.
2. SARS-CoV-2 DIPs, VLPs and Other Nanoparticles
Defective Viral Genomes (DVGs) can be formed during the replication of a virus when the polymerase switches between different templates or skips parts of the same template. Discontinuous transcription of CoV genomes enables recombination in a cell coinfected with more than one CoV species or variants via strand switching by the viral RdRp [11][12][13][14][15][16][17]. Some RNAs produced following such recombination procedures gather characteristics of DVGs including deletions that range from <1 kb to >20 kb; these DVGs retain intact 5′- and 3′-UTRs, and can be amplified by the CoV replication transcription complex (RTC) provided in trans by a helper virus. These recombination events have been attributed to the 3′–5′ exoribonuclease activity of the proofreading nsp14 protein and are responsible for CoV evolution. Packaging of DVGs into viral particles results in DIP production.
Another particle produced during CoV infections is the VLP. VLPs are self-assembled nanostructures composed of the structural proteins of a virus that, due to a lack of genetic material, are non-infectious. The data regarding the minimum requirements for the formation of SARS-CoV-2 VLPs and the ability of the membrane protein (M) alone to be secreted are controversial. However, co-expression of M with either the nucleocapsid (N) or the envelope (E) protein appears to be sufficient for VLP formation, while M, N and E together are required for optimal VLP production [18][19]. VLPs could advance our understanding of the assembly requirements for SARS-CoV-2, but could also be used for vaccine or interference strategies [20][21][22][23][24][25].
Finally, an analysis of plasma-derived nanoparticles from COVID-19 patients demonstrated that they were enriched in pro-inflammatory cytokines, IFN-γ, peptidases and proteases involved in vascular remodelling, and markers of cardiovascular tissue injury [26]. These nanoparticles could augment pro-inflammatory responses, endothelial dysfunction and thrombosis, which have been observed in severe COVID-19 cases.
3. Strategies
3.1. Targeting Macromolecular Assemblies
It has been persuasively argued that many cellular functions are performed by high-level complexes, modules or “supermolecules” [27][28]. Hence, those assemblies associated with viral processes can be targeted. The replication of coronaviruses involves the assembly of membrane-bound replicative organelles in which two lipid bilayers are closely paired [29]. This pairing is induced by the ribonucleoprotein complex proteins, which have been localized to networks of convoluted membranes and vesicles [30]. Targeting this network is important because viral polymerases that have a decreased fidelity often have an increased production of defective viral genomes [31][32][33]. Markers such as the Green Fluorescent Protein, chromobodies, or the haemagglutinin tag could be used to localize DVGs with the ability to encode proteins [34].
Poisoning complexes—and thereby disrupting functions—could be achieved in several ways. One way is via the production of novel or truncated peptides. Such production occurs in deletion DVGs derived from influenza [35]. DVGs constructed with mutations in the conserved regions, by which the influenza A polymerase subunits interact, inhibited polymerization and reduced virulence [36]. The multimerization of the polymerase is essential in the production of DVGs during influenza virus infection [32]. Such multimers might be perturbed either by altering the stoichiometry of coronavirus proteins or by generating incomplete RNAs or incomplete proteins that interfere with associations between macromolecules. For example, in the case of stoichiometry, this depends in part on post-translational regulation such as the phosphorylation of the serine and arginine residues in the SR-region of the SARS-CoV-2 N protein; a synDVG could therefore be created with this region deleted. In the case of incomplete proteins, deletion of the C-terminus of the N protein might disrupt the oligomerization of both the mutant and the wild-type protein. Sequestration of key constituents could be achieved by constructing synDVGs containing multiple copies of regulatory sequences, as illustrated by the sequestration of components of the Tat-based transcriptional activation system of HIV-1, where a vector was used that contained multiple copies of the sequences to which Tat binds [37]. Given the potential value of a recombinant protein, it may be worth considering the construction of a TIP to encode fusion of the ACE2 fragment to the M-protein to perturb SARS-CoV-2 assembly.
3.2. Targeting RNA
The transcription-regulating sequences (TRSs) at the 3′ end of the leader sequence (TRS-L) that precedes each viral gene (TRS-B) contains a conserved core sequence (CS) of 6–7 nucleotides along with variable 5′ and 3′ flanking sequences; as the conserved core sequence is identical for the genome leader (CS-L) and all mRNA coding sequences (CS-B), the CS-L may form a base-pair with the nascent negative strand complementary to each CS-B during the template-switching, which is central to transcription and replication [38]. The TRSs are therefore good candidates for targeting both viral transcription and replication.
3.3. Deletions
synDVGs constitute a powerful approach to viral therapy. Those that have deletions of part of the wt-genome could outcompete the wt-virus for the proteins and lipids essential to infectivity, which means wild-type virus release would be lower. Also, the synDVG could be released and transmitted to other cells where it could again hinder the replication of the wild-type virus. The factors to consider when designing synDVGs include the complex relationship between the length of the DVG, the degree of interference with the wild-type virus and the number of effective DVGs released. The minimum for a deletion DVG would be to have the 5′-UTR and 3′-UTR and the secondary structures they adopt to allow replication by the RdRp. Depending on whether the objective is to maximize production of DVGs or maximize interference, there is likely to be more than one optimum size. For example, the tight regulation of the quantity of each sub-genomic mRNA, which is believed to be important for the correct stoichiometry of the proteins of the wild-type virus, could be perturbed by a synDVG with partial deletions so that it encodes some proteins but not others.
The nature of the deletions in naturally occurring DIPs is useful in the design of synDVGs. An analysis of DVGs isolated from a respiratory syncytial virus indicated that the generation of copy-back mutations was not completely random but resulted from specific sequences encoded in the viral genome [39]. The analysis of the 5′ and 3′ regions flanking deletion sites during influenza infection was also consistent with conservation of specific sequences and structures [3][40].
A synthetic defective interfering SARS-CoV-2 was developed using the 5′ UTR and the adjacent 5′ part of nsp1 in ORF1a, the nsp15 that includes the putative packaging signal and the sequence spanning the 3′ part of the N sequence, ORF10 and the 3′-UTR. The rationale of this design is that a long ORF enables defective interfering genomes in some CoVs to replicate more efficiently and, since multiple transcriptional regulatory sequences (TRS) reduce replication efficiency, the 3′ portion was chosen to start within the N sequence to exclude its TRS. This synthetic defective genome was found to replicate three times faster than SARS-CoV-2 thereby reducing the viral load. Moreover, it transmitted as efficiently as the full-length genome, confirming the putative packaging signal of SARS-CoV-2 [41]. Based on this principle, two TIPs were developed recently that could inhibit SARS-CoV-2 in primary human lung organoids and in the Syrian Golden Hamster model of SARS-CoV-2 following intranasal delivery. These TIPs also reduced pro-inflammatory cytokines, and prevented pulmonary edema. The mechanism of SARS-CoV-2 inhibition by these TIPs was proposed to be due to competition for viral trans elements and no stimulation of innate immunity was recorded [42].
The use of a cocktail of different synDVGs directed against one or different targets may be more effective than a single synDVG. This is not only because synDVGs may act synergistically but also because the probability of the virus escaping inhibition by mutating is reduced. These “cocktail” synDVGs would be constructed so that they could neither complement one another nor recombine to generate the wild-type virus. In the case of HIV, antiviral genes were constructed that contained Tat- and Rev-binding decoys that acted synergistically [43], while a therapeutic vaccine, DermaVir, has been designed to boost T cell responses specific to 15 HIV antigens expressed from a single plasmid DNA [44][45].
Cocktails of synDVGs could have another advantage. There is no reason to suppose that viruses might cause only one pandemic at a time. We should therefore anticipate that two or more different viruses will eventually cause concurrent pandemics. Such a scenario could be dealt with by using a cocktail containing synDVGs to several different viruses. The success of such treatment would not depend on prior knowledge of the virus infecting or risking infecting a particular individual.
3.4. Immune Stimulation by TIPs
In designing an anti-viral therapy, one approach would be to construct a synDVG based on SARS-CoV-2 containing sequences resembling the copy-back sequences that stimulate the immune system. Alternatively, heterologous copy-back DVGs able to stimulate innate anti-viral immune responses strongly could be used. In the case of the Sendai virus (SeV), DVGs with copy-back genomes appear to be better at stimulating the immune system than those with a deleted genome [46]; although this was attributed to their long stretches of dsRNA, it has been argued that other characteristics of copy-back DVGs are also important contributors to the induction of anti-viral responses as shown by the induction of type 1 IFNs by the 44 nucleotide (nt)-long stem-loop motif in the copy-back genome of the DVG-546 Sendai virus [47]. SeV-based, copy-back DVGs increase the antigen presentation capacity of mouse and human dendritic cells, which increases the activation of T cells whilst, in the case of influenza A and respiratory syncytial virus, experimental vaccines with an adjuvant containing SeV-based, DVGs delivered subcutaneously, intramuscularly or intranasally have an increased level of antibodies and anti-viral protection [5][48][49][50].
A general purpose DVG might be developed based on the DIP 244 which was derived naturally from genome segment 1 of influenza A; this not only inhibits influenza viruses via RNA interference but also has a broad-spectrum activity against all other interferon-sensitive respiratory viruses via stimulation of type I interferon and pro-inflammatory cytokines [9][51][52][53]. It should be noted that SARS-CoV-2 is particularly sensitive to recombinant human IFN-α and IFN-β, which reduce viral titers [54]. Indeed, co-infections with IAV DIPs and SARS-CoV-2 led to abrogation of SARS-CoV-2 replication in a JAK/STAT-dependent mechanism [55]. Similarly, EVs from HSV-1 were found to restrict the respiratory syncytia virus (RSV). Thus, defective virus particles or other nanoparticles released from virus-infected cells could restrict heterologous viruses most likely through innate immunity activation [56].
3.5. Anti-Sense Oligonucleotides, Aptamers, Ribozymes, and Antibodies
Anti-sense RNA, RNA aptamers and ribozymes could all be incorporated into a TIP as could the RNA coding for the epitope-binding part of an antibody.
3.5.1. Anti-Sense Oligonucleotides
The anti-sense oligonucleotides (ASOs) strategy involves the use of nucleic acid strands of approximately 20 nt that can specifically hybridize to the complementary sequence of the target RNA [57][58]. The fate of the ASO:RNA hybrid varies depending on the ASO design strategy and either it could lead to cleavage of the mRNA, alter splicing, or it could form a steric blockade resulting in disruption of translation. Morpholino-type ASOs targeting the TRS in the 5′-UTR of the SARS-CoV block virus replication [59]. ASOs targeting conserved regions of the CoV genome such as the RdRp or the N sequence can be used to bypass issues of increased mutagenesis. An ASO-based strategy could be used to improve the effectiveness of treatments based on nucleoside analogues by splicing out ExoN [60].
Delivery of ASOs has been achieved using cationic polymers or by modifying them with lipids so they assemble in nanomicelles [61][62][63]. Phages are ideal vehicles for transferring nucleic acids, because they have the advantages of simple production, purification and a large capacity for containing genetic material. VLPs from bacteriophage Qβ have been used to encapsidate target RNAs to detect viral infections, including foot-and-mouth disease virus (FMDV) or Ebola virus. Asuragen and SeraCare have announced developments of SARS-CoV-2 positive controls for diagnostics, in which a SARS-CoV-2 detection module for RT-PCR was encapsidated into VLPs from the bacteriophage Qβ and the CCMV virus [64]. RNA nanoparticles of the bacteriophage φ29 have been used to deliver therapeutic oligonucleotides [65].
DNA rich in non-methylated CpG motifs are immunostimulatory and can be used as vaccine adjuvants or to stimulate protective immunity against pathogens. To enhance their stability and reduce serious side effects, a packaging and delivery strategy using VLPs has been proposed. These oligonucleotides induce protective cytotoxic T cell responses in the absence of systemic side-effects; hence, VLPs mounting protective immunity could accelerate SARS-CoV-2 clearance [66].
An alternative is to take a gene therapy approach, in which RNA oligos are encoded in a viral vector such as adeno-associated virus (AAV). Thus, short, hairpin RNAs can be fused to small PolIII promoters such as tRNA genes. The PolIII transcripts would be exported to the cytoplasm where the shRNAs would function in the RNAi pathway to knock down expression of various RNAs.
3.5.2. Aptamers
Nucleic acid aptamers are artificial, single-stranded or double-stranded DNA or RNA that can bind to their targets [67][68][69]. Because of their binding specificity, aptamers are often compared to antibodies [70][71]. Those that bind to viral proteins can have diagnostic and therapeutic potential. For example, aptamers binding to haemagglutinin have been used to detect different influenza strains [72]. Single-stranded DNA aptamers that bind to the ZIKA NS1 protein have diagnostic potential [73]. DNA aptamers against the dengue virus envelope protein can neutralize infection by all four serotypes of the virus [74]. RNA and DNA aptamers that disrupt the interaction of HSV-1 glycoprotein D with the virus entry receptor interfere with virus entry into the cells [75][76]. RNA aptamers were also described against the HIV-1 Gag protein that perturbed the Gag-genomic RNA interaction leading to the inhibition of HIV-1 genomic RNA levels [77]. Aptamers can also be used to suppress the activity of viral enzymes or host targets that contribute to pathogenesis [78][79]. Two RNA aptamers specific to the polymerase of HCV inhibited the initiation and the elongation of viral RNA synthesis by competing for the binding sites of the polymerase with viral RNA template [80].
RNA aptamers of approximately 40 nt were described for the SARS-CoV NTPase/helicase. The aptamers could inhibit the dsDNA unwinding activity of the helicase but not the ATPase [78]. A ssDNA aptamer that binds the N protein of SARS-CoV was proposed for diagnostic purposes [81]. This aptamer can also bind the N protein of SARS-CoV-2 [82]. Considering that the N protein is critical for nucleocapsid assembly, to antagonize host antiviral responses and the RNAi machinery, the aptamer targeting N protein could be repurposed to interfere with N functions [83][84]. Indeed, the C-terminus of the N protein contains a highly positively charged region possessing a strong affinity for ssDNA, ssRNA and dsDNA that can be easily targeted by aptamers. Also, an aptamer that binds nucleolin was found to inhibit SARS-CoV-2 replication [85]. Nucleolin is hijacked by the virus for its replication and the aptamer inhibits this process. Aptamers that bind the S protein have been designed mostly for diagnosis [86][87]. Aptamers that bind to the receptor-binding domain of S that could potentially inhibit virus entry were recently reported [88]. Aptamers that target the different methyltransferases of the virus, endonucleases and proteases are predicted to be effective.
Aptamers can be linked to therapeutic oligonucleotides such as siRNAs, miRNAs, and gRNAs forming chimeras that improve the properties of the therapeutic oligonucleotides [89]. RNA aptamer siRNA chimeric molecules could be designed to target the RNA genome/transcript of SARS-CoV-2.
3.5.3. Ribozymes and Antibodies
In the case of HIV, inactivation was achieved via anti-sense sequences against rev to prevent replication [90]: the Gag component of the capsid was fused to a calcium-sensitive nuclease to inactivate viral nucleic acids [91][92] and the 5′ leader was cleaved by a ribozyme [93]. These related technologies and DVGs based on SARS-CoV-2 could be used to target one or more of the RNA targets discussed above [94][95][96][97]. For example, the CRISPR/Cas13 RNA knockdown system delivered by the adeno-associated virus was used to cleave the SARS-CoV-2 RNA genome using guide RNAs to target the sequences encoding ORFab and the S-protein [98]. A single-domain camelid antibody against the S-protein can neutralize the SARS-CoV-2 pseudovirus [99]; such nanobodies could be encoded by a TIP.
3.6. EVs
EVs facilitate cell-to-cell communication and are produced by all types of cells. Recent studies demonstrated that COVID-19 patients had increased circulating platelet-derived EVs [100]. These EVs could transport platelet-derived cytokines and other proinflammatory molecules, including damage-associated molecular patterns [101]. The contribution of these EVs to COVID-19-associated coagulopathy and lung injury remains undetermined [102].
While EVs produced during an infection can exacerbate pathogenesis, those produced by uninfected cells could be modified and used for therapeutic purposes (Figure 1). EVs can be engineered to serve as carriers of nucleic acid sequences, including siRNAs, aptamers, genomes of TIPs, or other inhibitory molecules including proteins and small molecule inhibitors [103][104]. EVs would be more attractive if they could be directed to the desired targets. EVs produced by a particular type of cell, such as an immune cell, could target the proteins on the surface of diseased cells or an inflammatory immune cells [105]. Other strategies to redirect EVs include the expression of antibodies on the surface of EVs that target surface molecules in recipient cells or the expression of ligands to specific receptors.
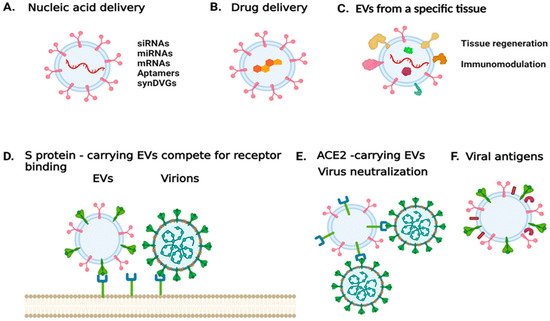
Figure 1. Harnessing EV properties to combat SARS-CoV-2 infection or treat COVID-19. (A) EVs can be used to deliver nucleic acid sequences that either modulate the expression of specific targets, express genes of interests, or encode for viral products. (B) EVs can be used to deliver compounds of interest. (C) EVs derived from a specific cell type or tissue could be used to mitigate disease and trigger tissue regeneration. (D) EVs carrying the spike protein can be used to antagonize viral entry into a host cell. (E) EVs carrying the virus entry receptor ACE2 could serve as decoys for the virus. (F) EVs carrying viral antigens could be used for vaccine development. The image was generated with BioRender.com (accessed on 13 September 2021).
In the case of SARS-CoV-2, vesicles could be designed to carry the S protein to antagonize virus entry [106]. Alternatively, EVs released during SARS-CoV-2 infection were found to carry ACE2; such EVs could be used to treat infections by coronaviruses that rely on ACE2 binding to enter host cells [107]. In addition, mesenchymal/stromal stem cell (MSC)-derived EVs have been shown to naturally target injured tissue and ameliorate acute organ injury [108]. These MSC EVs have been suggested for promoting recovery in patients with ARDS [109]. Thus, MSC EVs that carry ACE2 receptors could have a decoy function for SARS-CoV-2, while mitigating ARDS [110]. Currently, there are four clinical trials exploring the use of MSCEVs [111]. One trial aims to determine the therapeutic potential of aerosol inhalation of adipose-tissue derived MSC EVs in patients with severe COVID-19. The safety profile of this treatment is assessed in a different trial. An additional trial aims to determine whether MSC EVs can suppress immune system over-response to the virus, and whether they can trigger regenerative processes. In other trials, bone marrow-derived MSC EVs are being tested in COVID-19 patients with moderate-to-severe ARDS [111].
Finally, viral antigens on EVs could also be used for vaccine development or to serve as adjuvants. Depending on the status and the type of cell from which they originated, EVs may facilitate the initiation, expansion, maintenance, or silencing of adaptive immune responses [112].
3.7. Mimics
One possibility would be to produce the ACE2 receptor fragment, to which SARS-CoV-2 binds, on the surface of a bacteriophage or a bacterium to act as a competitor; these phages or bacteria might then be used to impregnate masks and coat other surfaces, including skin and mucosal membranes, or even be used as inhalants. There is a novel technique that could be used to produce peptides that would bind proteins on the surface of the virus and, indeed, that would bind the other viral proteins (and that could then be encoded by a synDVG). This technique is the Mimic Chain Reaction [113] which, in a sense, is the peptide equivalent of the PCR technique. The Mimic Chain Reaction is based on the auto-induction or quorum-sensing systems of bacteria and allows both the selection of peptides that bind to a target and peptides that mimic the epitopes of the target.
References
- Ganem, D.E. 2000 and Beyond: Confronting the Microbe Menace Lecture One—Microbe Hunters: Tracking Infectious Agents. Available online: https://www.biointeractive.org/sites/default/files/Infectious%2520Diseases%2520Lecture%25201%2520Transcript.pdf (accessed on 9 June 2020).
- Noppornpanth, S.; Smits, S.L.; Lien, T.X.; Poovorawan, Y.; Osterhaus, A.D.; Haagmans, B.L. Characterization of hepatitis c virus deletion mutants circulating in chronically infected patients. J. Virol. 2007, 81, 12496–12503.
- Saira, K.; Lin, X.; DePasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence analysis of in vivo defective interfering-like rna of influenza a h1n1 pandemic virus. J. Virol. 2013, 87, 8064–8074.
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri, R.A., Jr.; Hodinka, R.L.; Lopez, C.B. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog. 2015, 11, e1005122.
- Vignuzzi, M.; Lopez, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087.
- Roux, L.; Simon, A.E.; Holland, J.J. Effects of defective interfering viruses on virus-replication and pathogenesis em in vitro and em in vivo. Adv. Virus Res. 1991, 40, 181–211.
- Laske, T.; Heldt, F.S.; Hoffmann, H.; Frensing, T.; Reichl, U. Modeling the intracellular replication of influenza a virus in the presence of defective interfering rnas. Virus Res. 2016, 213, 90–99.
- Duhaut, S.D.; McCauley, J.W. Defective rnas inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology 1996, 216, 326–337.
- Easton, A.J.; Scott, P.D.; Edworthy, N.L.; Meng, B.; Marriott, A.C.; Dimmock, N.J. A novel broad-spectrum treatment for respiratory virus infections: Influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 2011, 29, 2777–2784.
- Notton, T.; Sardanyes, J.; Weinberger, A.D.; Weinberger, L.S. The case for transmissible antivirals to control population-wide infectious disease. Trends Biotechnol. 2014, 32, 400–405.
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between nonsegmented rna genomes of murine coronaviruses. J. Virol. 1985, 56, 449–456.
- Makino, S.; Stohlman, S.A.; Lai, M.M. Leader sequences of murine coronavirus mrnas can be freely reassorted: Evidence for the role of free leader rna in transcription. Proc. Natl. Acad. Sci. USA 1986, 83, 4204–4208.
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-frequency rna recombination of murine coronaviruses. J. Virol. 1986, 57, 729–737.
- Lai, M.M.; Makino, S.; Soe, L.H.; Shieh, C.K.; Keck, J.G.; Fleming, J.O. Coronavirus: A jumping rna transcription. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 359–365.
- Lai, M.M. Molecular biology of coronavirus 1986. Adv. Exp. Med. Biol. 1987, 218, 7–13.
- Keck, J.G.; Soe, L.H.; Makino, S.; Stohlman, S.A.; Lai, M.M. Rna recombination of murine coronaviruses: Recombination between fusion-positive mouse hepatitis virus a59 and fusion-negative mouse hepatitis virus 2. J. Virol. 1988, 62, 1989–1998.
- Keck, J.G.; Matsushima, G.K.; Makino, S.; Fleming, J.O.; Vannier, D.M.; Stohlman, S.A.; Lai, M.M. In vivo rna-rna recombination of coronavirus in mouse brain. J. Virol. 1988, 62, 1810–1813.
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 virus-like particles by mammalian expression system. Front. Bioeng. Biotechnol. 2020, 8, 862.
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 viral budding and entry can be modeled using bsl-2 level virus-like particles. J. Biol. Chem. 2021, 296, 100103.
- Kuo, L.; Masters, P.S. The small envelope protein e is not essential for murine coronavirus replication. J. Virol. 2003, 77, 4597–4608.
- Ortego, J.; Escors, D.; Laude, H.; Enjuanes, L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002, 76, 11518–11529.
- Ortego, J.; Ceriani, J.E.; Patino, C.; Plana, J.; Enjuanes, L. Absence of e protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308.
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the e gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713.
- Dediego, M.L.; Pewe, L.; Alvarez, E.; Rejas, M.T.; Perlman, S.; Enjuanes, L. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hace-2 transgenic mice. Virology 2008, 376, 379–389.
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194, 124–137.
- Krishnamachary, B.; Cook, C.; Spikes, L.; Chalise, P.; Dhillon, N.K. The potential role of extracellular vesicles in COVID-19 associated endothelial injury and pro-inflammation. medRxiv 2020.
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52.
- Schubert, W.; Gieseler, A.; Krusche, A.; Serocka, P.; Hillert, R. Next-generation biomarkers based on 100-parameter functional super-resolution microscopy tis. New Biotechnol. 2012, 29, 599–610.
- V’Kovski, P.; Al-Mulla, H.; Thiel, V.; Neuman, B.W. New insights on the role of paired membrane structures in coronavirus replication. Virus Res. 2015, 202, 33–40.
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19.
- Fodor, E.; Mingay, L.J.; Crow, M.; Deng, T.; Brownlee, G.G. A single amino acid mutation in the pa subunit of the influenza virus rna polymerase promotes the generation of defective interfering rnas. J. Virol. 2003, 77, 5017–5020.
- Vasilijevic, J.; Zamarreno, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gomez, G.; Rodriguez, G.; Perez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650.
- Poirier, E.Z.; Mounce, B.C.; Rozen-Gagnon, K.; Hooikaas, P.J.; Stapleford, K.A.; Moratorio, G.; Vignuzzi, M. Low-fidelity polymerases of alphaviruses recombine at higher rates to overproduce defective interfering particles. J. Virol. 2015, 90, 2446–2454.
- Zeng, L.P.; Gao, Y.T.; Ge, X.Y.; Zhang, Q.; Peng, C.; Yang, X.L.; Tan, B.; Chen, J.; Chmura, A.A.; Daszak, P.; et al. Bat severe acute respiratory syndrome-like coronavirus wiv1 encodes an extra accessory protein, orfx, involved in modulation of the host immune response. J. Virol. 2016, 90, 6573–6582.
- Akkina, R.K.; Chambers, T.M.; Nayak, D.P. Expression of defective-interfering influenza virus-specific transcripts and polypeptides in infected cells. J. Virol. 1984, 51, 395–403.
- Manz, B.; Gotz, V.; Wunderlich, K.; Eisel, J.; Kirchmair, J.; Stech, J.; Stech, O.; Chase, G.; Frank, R.; Schwemmle, M. Disruption of the viral polymerase complex assembly as a novel approach to attenuate influenza a virus. J. Biol. Chem. 2011, 286, 8414–8424.
- Lisziewicz, J.; Rappaport, J.; Dhar, R. Tat-regulated production of multimerized tar rna inhibits hiv-1 gene expression. New Biol. 1991, 3, 82–89.
- Sola, I.; Almazan, F.; Zuniga, S.; Enjuanes, L. Continuous and discontinuous rna synthesis in coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288.
- Sun, Y.; Kim, E.J.; Felt, S.A.; Taylor, L.J.; Agarwal, D.; Grant, G.R.; Lopez, C.B. A specific sequence in the genome of respiratory syncytial virus regulates the generation of copy-back defective viral genomes. PLoS Pathog. 2019, 15, e1007707.
- Jennings, P.A.; Finch, J.T.; Winter, G.; Robertson, J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral rna? Cell 1983, 34, 619–627.
- Yao, S.; Narayanan, A.; Majowicz, S.A.; Jose, J.; Archetti, M. A synthetic defective interfering SARS-CoV-2. PeerJ 2021, 9, e11686.
- Chaturvedi, S.; Vasen, G.; Pablo, M.; Chen, X.; Beutler, N.; Kumar, A.; Tanner, E.; Illouz, S.; Rahgoshay, D.; Burnett, J.; et al. Identification of a therapeutic interfering particle-a single-dose SARS-CoV-2 antiviral intervention with a high barrier to resistance. Cell 2021, 184, 6022–6036.e18.
- Lisziewicz, J.; Zeng, G.; Gratas, C.; Weinstein, J.N.; Lori, F. Combination gene therapy: Synergistic inhibition of human immunodeficiency virus tat and rev functions by a single rna molecule. Hum. Gene 2000, 11, 807–815.
- Somogyi, E.; Xu, J.; Gudics, A.; Toth, J.; Kovacs, A.L.; Lori, F.; Lisziewicz, J. A plasmid DNA immunogen expressing fifteen protein antigens and complex virus-like particles (vlp+) mimicking naturally occurring hiv. Vaccine 2011, 29, 744–753.
- DermaVir. Available online: http://www.geneticimmunity.com/dermavir.html (accessed on 13 January 2022).
- Strahle, L.; Garcin, D.; Kolakofsky, D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 2006, 351, 101–111.
- Xu, J.; Mercado-Lopez, X.; Grier, J.T.; Kim, W.K.; Chun, L.F.; Irvine, E.B.; Del Toro Duany, Y.; Kell, A.; Hur, S.; Gale, M., Jr.; et al. Identification of a natural viral rna motif that optimizes sensing of viral rna by rig-i. mBio 2015, 6, e01265-15.
- Mercado-Lopez, X.; Cotter, C.R.; Kim, W.K.; Sun, Y.; Munoz, L.; Tapia, K.; Lopez, C.B. Highly immunostimulatory rna derived from a sendai virus defective viral genome. Vaccine 2013, 31, 5713–5721.
- Martinez-Gil, L.; Goff, P.H.; Hai, R.; Garcia-Sastre, A.; Shaw, M.L.; Palese, P. A sendai virus-derived rna agonist of rig-i as a virus vaccine adjuvant. J. Virol. 2013, 87, 1290–1300.
- Fisher, D.G.; Coppock, G.M.; Lopez, C.B. Virus-derived immunostimulatory rna induces type i ifn-dependent antibodies and t-cell responses during vaccination. Vaccine 2018, 36, 4039–4045.
- Dimmock, N.J.; Rainsford, E.W.; Scott, P.D.; Marriott, A.C. Influenza virus protecting rna: An effective prophylactic and therapeutic antiviral. J. Virol. 2008, 82, 8570–8578.
- Meng, B.; Bentley, K.; Marriott, A.C.; Scott, P.D.; Dimmock, N.J.; Easton, A.J. Unexpected complexity in the interference activity of a cloned influenza defective interfering rna. Virol. J. 2017, 14, 138.
- Smith, C.M.; Scott, P.D.; O’Callaghan, C.; Easton, A.J.; Dimmock, N.J. A defective interfering influenza rna inhibits infectious influenza virus replication in human respiratory tract cells: A potential new human antiviral. Viruses 2016, 8, 237.
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type i interferons to SARS-CoV-2 infection. Antivir. Res 2020, 179, 104811.
- Rand, U.; Kupke, S.Y.; Shkarlet, H.; Hein, M.D.; Hirsch, T.; Marichal-Gallardo, P.; Cicin-Sain, L.; Reichl, U.; Bruder, D. Antiviral activity of influenza a virus defective interfering particles against SARS-CoV-2 replication in vitro through stimulation of innate immunity. Cells 2021, 10, 1756.
- Dogrammatzis, C.; Saleh, S.; Deighan, C.; Kalamvoki, M. Diverse populations of extracellular vesicles with opposite functions during herpes simplex virus 1 infection. J. Virol. 2021, 95, e02357-20.
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus rna proofreading: Molecular basis and therapeutic targeting. Mol. Cell 2020, 79, 710–727.
- Gagliardi, M.; Ashizawa, A.T. The challenges and strategies of antisense oligonucleotide drug delivery. Biomedicines 2021, 9, 433.
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Churchill, M.J.; Kim, A.M.; Kuhn, P.; Dawson, P.; Moulton, H.M.; Bestwick, R.K.; Iversen, P.L.; et al. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 2005, 79, 9665–9676.
- Shimo, T.; Maruyama, R.; Yokota, T. Designing effective antisense oligonucleotides for exon skipping. Methods Mol. Biol. 2018, 1687, 143–155.
- Karaki, S.; Benizri, S.; Mejias, R.; Baylot, V.; Branger, N.; Nguyen, T.; Vialet, B.; Oumzil, K.; Barthelemy, P.; Rocchi, P. Lipid-oligonucleotide conjugates improve cellular uptake and efficiency of tctp-antisense in castration-resistant prostate cancer. J. Control Release 2017, 258, 1–9.
- Posocco, P.; Liu, X.; Laurini, E.; Marson, D.; Chen, C.; Liu, C.; Fermeglia, M.; Rocchi, P.; Pricl, S.; Peng, L. Impact of sirna overhangs for dendrimer-mediated sirna delivery and gene silencing. Mol. Pharm. 2013, 10, 3262–3273.
- Luvino, D.; Khiati, S.; Oumzil, K.; Rocchi, P.; Camplo, M.; Barthelemy, P. Efficient delivery of therapeutic small nucleic acids to prostate cancer cells using ketal nucleoside lipid nanoparticles. J. Control Release 2013, 172, 954–961.
- Chan, S.K.; Du, P.; Ignacio, K.; Metha, S.; Newton, I.G.; Steinmetz, N.F. Biomimetic virus-like particles as SARS-CoV-2 positive controls for rt-pcr diagnostics. medRxiv 2021, 15, 1259–1272.
- Karimi, M.; Mirshekari, H.; Moosavi Basri, S.M.; Bahrami, S.; Moghoofei, M.; Hamblin, M.R. Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 2016, 106, 45–62.
- Storni, T.; Ruedl, C.; Schwarz, K.; Schwendener, R.A.; Renner, W.A.; Bachmann, M.F. Nonmethylated cg motifs packaged into virus-like particles induce protective cytotoxic t cell responses in the absence of systemic side effects. J. Immunol. 2004, 172, 1777–1785.
- Schneider, D.; Tuerk, C.; Gold, L. Selection of high affinity rna ligands to the bacteriophage r17 coat protein. J. Mol. Biol. 1992, 228, 862–869.
- Torres-Chavolla, E.; Alocilja, E.C. Aptasensors for detection of microbial and viral pathogens. Biosens. Bioelectron. 2009, 24, 3175–3182.
- Ku, T.H.; Zhang, T.; Luo, H.; Yen, T.M.; Chen, P.W.; Han, Y.; Lo, Y.H. Nucleic acid aptamers: An emerging tool for biotechnology and biomedical sensing. Sensors 2015, 15, 16281–16313.
- Banerjee, J. Antibodies are challenged. Indian J. Med. Sci. 2010, 64, 144–147.
- Szpechcinski, A.; Grzanka, A. Aptamers in clinical diagnostics. Postepy Biochem. 2006, 52, 260–270.
- Zou, X.; Wu, J.; Gu, J.; Shen, L.; Mao, L. Application of aptamers in virus detection and antiviral therapy. Front. Microbiol. 2019, 10, 1462.
- Lee, K.H.; Zeng, H. Aptamer-based elisa assay for highly specific and sensitive detection of zika ns1 protein. Anal. Chem. 2017, 89, 12743–12748.
- Chen, H.L.; Hsiao, W.H.; Lee, H.C.; Wu, S.C.; Cheng, J.W. Selection and characterization of DNA aptamers targeting all four serotypes of dengue viruses. PLoS ONE 2015, 10, e0131240.
- Gopinath, S.C.; Hayashi, K.; Kumar, P.K. Aptamer that binds to the gd protein of herpes simplex virus 1 and efficiently inhibits viral entry. J. Virol. 2012, 86, 6732–6744.
- Yadavalli, T.; Agelidis, A.; Jaishankar, D.; Mangano, K.; Thakkar, N.; Penmetcha, K.; Shukla, D. Targeting herpes simplex virus-1 gd by a DNA aptamer can be an effective new strategy to curb viral infection. Mol. Nucleic Acids 2017, 9, 365–378.
- Ramalingam, D.; Duclair, S.; Datta, S.A.; Ellington, A.; Rein, A.; Prasad, V.R. Rna aptamers directed to human immunodeficiency virus type 1 gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J. Virol. 2011, 85, 305–314.
- Jang, K.J.; Lee, N.R.; Yeo, W.S.; Jeong, Y.J.; Kim, D.E. Isolation of inhibitory rna aptamers against severe acute respiratory syndrome (sars) coronavirus ntpase/helicase. Biochem. Biophys. Res. Commun. 2008, 366, 738–744.
- Shum, K.T.; Tanner, J.A. Differential inhibitory activities and stabilisation of DNA aptamers against the sars coronavirus helicase. Chembiochem 2008, 9, 3037–3045.
- Bellecave, P.; Cazenave, C.; Rumi, J.; Staedel, C.; Cosnefroy, O.; Andreola, M.L.; Ventura, M.; Tarrago-Litvak, L.; Astier-Gin, T. Inhibition of hepatitis c virus (hcv) rna polymerase by DNA aptamers: Mechanism of inhibition of in vitro rna synthesis and effect on hcv-infected cells. Antimicrob. Agents Chemother. 2008, 52, 2097–2110.
- Cho, S.J.; Woo, H.M.; Kim, K.S.; Oh, J.W.; Jeong, Y.J. Novel system for detecting sars coronavirus nucleocapsid protein using an ssdna aptamer. J. Biosci. Bioeng. 2011, 112, 535–540.
- Chen, Z.; Wu, Q.; Chen, J.; Ni, X.; Dai, J. A DNA aptamer based method for detection of SARS-CoV-2 nucleocapsid protein. Virol. Sin. 2020, 35, 351–354.
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018.
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The nucleocapsid protein of coronaviruses acts as a viral suppressor of rna silencing in mammalian cells. J. Virol. 2015, 89, 9029–9043.
- Wang, Y.L.; McKeague, M. Aptamers in the pursuit of COVID-19 management. Aptamers 2020, 4, 1–2.
- Liebich, S. Potent selex aptamer-based therapeutic method for novel SARS-CoV2 virus disease (COVID-19). COVID-19 BabuBio 2020, 53, 101636.
- Rangan, R.; Watkins, A.M.; Chacon, J.; Kladwang, W.; Zheludev, I.N.; Townley, J.; Rynge, M.; Thain, G.; Das, R. De novo 3d models of SARS-CoV-2 rna elements and small-molecule-binding rnas to aid drug discovery. bioRxiv 2020, 49, 3092–3108.
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900.
- Zhou, J.; Rossi, J.J. Aptamer-targeted rnai for hiv-1 therapy. Methods Mol. Biol. 2011, 721, 355–371.
- Matsukura, M.; Zon, G.; Shinozuka, K.; Robert-Guroff, M.; Shimada, T.; Stein, C.A.; Mitsuya, H.; Wong-Staal, F.; Cohen, J.S.; Broder, S. Regulation of viral expression of human immunodeficiency virus in vitro by an antisense phosphorothioate oligodeoxynucleotide against rev (art/trs) in chronically infected cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4244–4248.
- Natsoulis, G.; Boeke, J.D. New antiviral strategy using capsid-nuclease fusion proteins. Nature 1991, 352, 632–635.
- Natsoulis, G.; Seshaiah, P.; Federspiel, M.J.; Rein, A.; Hughes, S.H.; Boeke, J.D. Targeting of a nuclease to murine leukemia virus capsids inhibits viral multiplication. Proc. Natl. Acad. Sci. USA 1995, 92, 364–368.
- Yu, M.; Ojwang, J.; Yamada, O.; Hampel, A.; Rapapport, J.; Looney, D.; Wong-Staal, F. A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1993, 90, 6340–6344.
- Asha, K.; Kumar, P.; Sanicas, M.; Meseko, C.A.; Khanna, M.; Kumar, B. Advancements in nucleic acid based therapeutics against respiratory viral infections. J. Clin. Med. 2018, 8, 6.
- Goguen, R.P.; Malard, C.M.; Scarborough, R.J.; Gatignol, A. Small rnas to treat human immunodeficiency virus type 1 infection by gene therapy. Curr. Opin. Virol. 2019, 38, 10–20.
- Nguyen, P.D.M.; Zheng, J.; Gremminger, T.J.; Qiu, L.; Zhang, D.; Tuske, S.; Lange, M.J.; Griffin, P.R.; Arnold, E.; Chen, S.J.; et al. Binding interface and impact on protease cleavage for an rna aptamer to hiv-1 reverse transcriptase. Nucleic Acids Res. 2020, 48, 2709–2722.
- Zhong, G.; Wang, H.; He, W.; Li, Y.; Mou, H.; Tickner, Z.J.; Tran, M.H.; Ou, T.; Yin, Y.; Diao, H.; et al. A reversible rna on-switch that controls gene expression of aav-delivered therapeutics in vivo. Nat. Biotechnol. 2020, 38, 169–175.
- Nguyen, T.M.; Zhang, Y.; Pandolfi, P.P. Virus against virus: A potential treatment for 2019-ncov (SARS-CoV-2) and other rna viruses. Cell Res. 2020, 30, 189–190.
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Team, V.-C.C.-R.; Hoffmann, M.; et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020, 181, 1004–1015.e1015.
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating platelet-derived extracellular vesicles are a hallmark of SARS-CoV-2 infection. Cells 2021, 10, 85.
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets can associate with SARS-CoV-2 rna and are hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418.
- Traby, L.; Kollars, M.; Kussmann, M.; Karer, M.; Sinkovec, H.; Lobmeyr, E.; Hermann, A.; Staudinger, T.; Schellongowski, P.; Rossler, B.; et al. Extracellular vesicles and citrullinated histone h3 in coronavirus disease 2019 patients. Thromb. Haemost. 2021.
- Kumar, L.; Verma, S.; Vaidya, B.; Gupta, V. Exosomes: Natural carriers for sirna delivery. Curr. Pharm. Des. 2015, 21, 4556–4565.
- Kumar, S.; Zhi, K.; Mukherji, A.; Gerth, K. Repurposing antiviral protease inhibitors using extracellular vesicles for potential therapy of COVID-19. Viruses 2020, 12, 486.
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494.
- Vallhov, H.; Gutzeit, C.; Johansson, S.M.; Nagy, N.; Paul, M.; Li, Q.; Friend, S.; George, T.C.; Klein, E.; Scheynius, A.; et al. Exosomes containing glycoprotein 350 released by ebv-transformed b cells selectively target b cells through cd21 and block ebv infection in vitro. J. Immunol. 2011, 186, 73–82.
- El-Shennawy, L.; Hoffmann, A.D.; Dashzeveg, N.K.; Mehl, P.J.; Yu, Z.; Tokars, V.L.; Nicolaescu, V.; Ostiguin, C.; Jia, Y.; Li, L.; et al. Circulating ace2-expressing exosomes block SARS-CoV-2 infection as an innate antiviral mechanism. bioRxiv 2020.
- Park, K.S.; Bandeira, E.; Shelke, G.V.; Lasser, C.; Lotvall, J. Enhancement of therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res. Ther. 2019, 10, 288.
- Shah, T.; Qin, S.; Vashi, M.; Predescu, D.N.; Jeganathan, N.; Bardita, C.; Ganesh, B.; diBartolo, S.; Fogg, L.F.; Balk, R.A.; et al. Alk5/runx1 signaling mediated by extracellular vesicles promotes vascular repair in acute respiratory distress syndrome. Clin. Transl. Med. 2018, 7, 19.
- Inal, J.M. Decoy ace2-expressing extracellular vesicles that competitively bind SARS-CoV-2 as a possible COVID-19 therapy. Clin. Sci. 2020, 134, 1301–1304.
- Lee, B.C.; Kang, I.; Yu, K.R. Therapeutic features and updated clinical trials of mesenchymal stem cell (msc)-derived exosomes. J. Clin. Med. 2021, 10, 711.
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu. Rev. Immunol. 2018, 36, 435–459.
- Norris, V.; Thierry, A.; Holland, I.B.; Amar, P.; Molina, F. The mimic chain reaction. J. Mol. Microbiol. Biotechnol. 2012, 22, 335–343.
More
Information
Subjects:
Virology; Infectious Diseases
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
28 Jan 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No