Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Ilari | + 4863 word(s) | 4863 | 2021-12-21 07:50:47 | | | |
| 2 | Camila Xu | -2291 word(s) | 2572 | 2022-01-27 02:34:23 | | | | |
| 3 | Camila Xu | -2291 word(s) | 2572 | 2022-01-27 02:35:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ilari, A. Huntingtin Ubiquitination Mechanisms. Encyclopedia. Available online: https://encyclopedia.pub/entry/18837 (accessed on 25 July 2026).
Ilari A. Huntingtin Ubiquitination Mechanisms. Encyclopedia. Available at: https://encyclopedia.pub/entry/18837. Accessed July 25, 2026.
Ilari, Andrea. "Huntingtin Ubiquitination Mechanisms" Encyclopedia, https://encyclopedia.pub/entry/18837 (accessed July 25, 2026).
Ilari, A. (2022, January 26). Huntingtin Ubiquitination Mechanisms. In Encyclopedia. https://encyclopedia.pub/entry/18837
Ilari, Andrea. "Huntingtin Ubiquitination Mechanisms." Encyclopedia. Web. 26 January, 2022.
Copy Citation
Huntington Disease (HD) is caused by the CAG repeat expansion (≥36 CAG triplets) in the exon1 of the HTT gene encoding for the protein huntingtin (Htt). Huntingtin and mutated huntingtin (mHtt) are degradated by the ubiquitin-proteasome system (UPS). Ubiquitination has been linked to reduced mHtt toxicity, most likely due to increased mHtt clearance by the proteasome.
ubiquitination
huntingtin
E3 ligases
1. Introduction
Huntington disease (HD) is an autosomal, dominant, lethal neurodegenerative disorder affecting between 0.42 and 17.2 per 100,000 individuals around the world [1][2][3] HD results in a wide range of symptoms, including involuntary movements, clumsiness, lack of concentration, memory lapses, mood swings, and depression. Although brain pathology is considered a hallmark of HD, new studies suggest that peripheral tissue pathology is an important factor in disease manifestation and progression. In particular, HD mouse models have recently been shown to display skeletal muscle malfunction and HD-related cardiomyopathy [4][5][6].
Primary symptoms generally occur in adults (typical age range: 40–45 years) and gradually progress, leading to the deterioration of patients’ health, and finally death, after 10–20 years. Although several clinical trials are ongoing, there is no cure for HD at present. HD is caused by a mutation in the HTT gene, which encodes for the huntingtin (Htt) protein. The HTT gene contains a repeat of 6–35 CAG triplets in exon 1, which is translated into a polyglutamine (polyQ) stretch in the Htt N-terminal region. The HD patients’ gene contains ≥36 CAG triplets, encoding for a mutated Htt (mHtt) with an expanded polyQ region [7]. mHtt is highly susceptible to aggregation with other mHtt molecules or different proteins, leading to the formation of clusters, fibrils and inclusions, some of which comprise 100,000s of mHtt molecules and are large enough to be visualized by light microscopy [8][9].
CAG stretches that are longer than 60 repeats in the Htt gene are associated with Juvenile-onset Huntington Disease (JoHD). This is a rare HD variant that accounts for about 4–10% of all cases, typically defined based on the appearance of symptoms at age 20 years or younger [10]. JoHD patients experience different motor and non-motor symptoms at disease onset and throughout the disease course, with a faster disease progression rate and reduced life span with respect to adult HD patients [11].
Since both soluble and aggregated mHtt are well known to induce ER (Endoplasmic Reticulum) stress, leading to neuronal injury and apoptosis [12], both the inhibition of mHtt aggregate formation and the acceleration of mHtt degradation could be exploited for HD symptom delay, or even treatment [13]. mHtt can be degraded by two main pathways: the ubiquitin–proteasome system (UPS) and the autophagy–lysosomal pathway. In this paper, we focus on the UPS, since it has been shown to play a more important role in removing mHtt than the autophagy–lysosomal pathway [14]. In this framework, it is important to underline that cells contain a variety of molecular chaperones and other proteins, such as heat shock family proteins HSP40, HSP70, HSP90, and HSP105, which are able to identify and combine with misfolded mHtt to inhibit aggregate formation to different degrees, leading to cell survival [15][16][17][18][19].
Here, we describe the mechanism of mHtt ubiquitination, with particular emphasis on the structure–function relationships in E3 ligases involved in the process. Finally, we discuss how knowledge of these relationships can be exploited to develop PROteolysis TArgeting Chimeras (PROTACs), which may be used to develop innovative drugs able to increase mHtt clearance in HD patients.
2. Ubiquitin Proteasome System (UPS)
The UPS comprises ubiquitin (Ub), proteasome, and three classes of enzymes, and plays key roles in various essential biological processes, such as cell cycle progression, signal transduction, maintenance of genome integrity, and tumorigenesis [20].
UPS activity consists of two main processes: (1) covalent attachment of multiple Ub molecules to the target protein; and (2) degradation of the resulting covalent target protein–Ub complex by the 26S proteasome complex (Figure 1). The first process, in turn, encompasses several steps and requires at least three classes of enzymes: (i) Ub-activating enzymes (E1); (ii) Ub-conjugating enzymes (E2); and (iii) Ub ligases (E3). In the first step, E1 enzymes use ATP to activate the Ub carboxy-terminal region (C-ter). The resulting Ub–AMP drives the formation of a thioester bond between Ub and a cysteine residue of E1. In the second step, Ub is transferred to a cysteine residue of an E2 enzyme. In the third step, the E3 ligase binds both the Ub–E2 adduct and the target protein. In step four, an isopeptide bond between Ub and the protein target is formed (Figure 1) [21][22]. The process that consists in the linkage of a protein to a single Ub monomer is known as monoubiquitination.
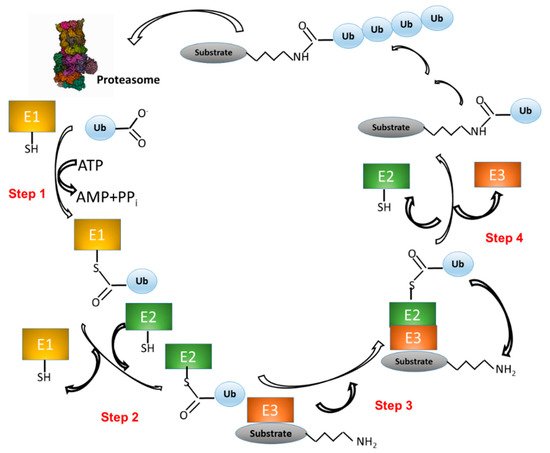
Figure 1. The ubiquitination pathway. (1) Ubiquitin (Ub) is linked to a cysteine residue of a Ub-activating enzyme (E1) via a reaction that uses the energy derived from the hydrolysis of an ATP molecule. (2) Ub is transferred from E1 to the cysteine of a Ub-conjugating enzyme (E2). (3) A complex is formed between E2–Ub and a Ub ligase (E3), which is able to bind a protein substrate. (4) Ub is transferred from E2 to a lysine residue of the protein substrate. Other Ub molecules can be added to the substrate–Ub complex with the same mechanism (steps 1–4). Finally, the polyubiquitinated substrate is recognized and degraded by the proteasome, whereas Ub is released and ready to start a new cycle.
Once monoubiquitination has occurred, the Ub moiety can be ubiquitinated again at one of the seven lysine sidechains (K6, K11, K27, K29, K33, K48, and K63) or at the free main-chain amine group of the N-terminal methionine residue (M1) (Figure 2), thus extending the modification into a polyubiquitin chain. The ability of Ub to link other Ub molecules at eight different sites is the molecular basis of a dynamic and complex system regulating cellular metabolism. In turn, the different chain types resulting from polyubiquitination processes dictate the specific signaling function associated with the modification [23][24][25].
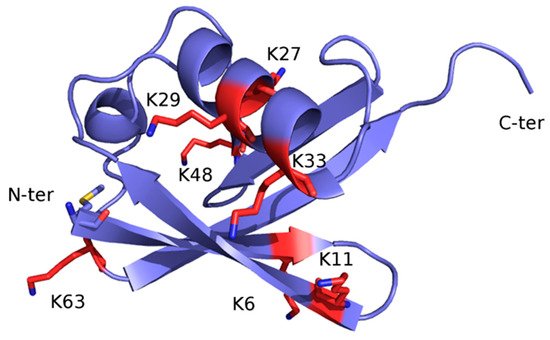
Figure 2. X-ray structure of Ub (PDB code: 1UBQ). The structure is represented as ribbon and colored light blue. The seven lysine residues are represented as sticks and colored red, except for the side chain amino group, which is blue. The N-terminal methionine residue (M1) is shown as sticks and colored by atom type: blue, N (of the main-chain free amino group); red, O; yellow, S; light blue, C.
Polyubiquitin linkages are classified as: (i) homotypic, if Ub molecules are bound to a single lysine position of the substrate; (ii) heterotypic, in case Ub molecules are bound at different lysine positions of the substrate; (iii) branched, when ubiquitination takes place at two or more sites on a single Ub molecule [26]. Ub exerts several specific functions, which are contributed not only by monoubiquitin, but also by numerous different combinations of polyubiquitin linkages.
Monoubiquitination contributes to several processes, including DNA repair, control of transcription, metabolism, and apoptosis [27].
The role of homotypic chains depends on the position of the lysine residue involved in the linkage. K48 Ub, wherein a Ub chain is covalently bonded to the ε-amino group of the lysine at position 48 of the preceding Ub molecule, is the most abundant linkage type in homotypic polyubiquitin chains and represents the canonical signal for proteasomal degradation [28]. K11 Ub is particularly abundant as well [26] and has proteasome-independent functions, including intracellular signaling [29][30], in addition to proteasomal-dependent degradation [31]. K63 Ub regulates several processes, including the multiple translation process independent of the proteasome. This encompasses translation quality control, in particular during oxidative stress, and is also known to induce autophagy [27][32][33].
The heterotypic, branched K48 and K11 are associated with proteasomal degradation of several cell-cycle regulators, including cyclin B1 and securin, thereby promoting mitotic exit.
3. The Level of Huntingtin Is Controlled by Ubiquitination
3.1. Htt Structure and Function
In HD patients, the polyQ region located after the first seventeen N-terminal Htt residues is expanded beyond a threshold of 36 glutamine residues (mHtt) [34].
A distinctive feature of HD is the progressive death of striatal projection neurons (SPNs), which are GABAergic output neurons representing >90% of striatal cells. SPNs are divided into two groups, depending on whether they belong to the direct (DP) or indirect (IP) pathway (DP-SPNs and IP-SPNs, respectively). Both SPN sub-types receive extensive glutamatergic inputs from cortex and thalamus, and dopaminergic inputs from the ventral tegmental area and substantia nigra pars compacta [34].
Within neurons, mHtt molecules form toxic aggregates with a rate proportional to the length of the polyQ expansion. mHtt co-aggregates with a number of different proteins, which decreases their concentration in the cell. Several studies demonstrate that polyQ expansion in mHtt results in a toxic gain-of-function phenotype. Other studies, which include gene knockouts and knockdowns, demonstrate that polyQ expansion in mHtt can also have loss-of-function effects. The role of wild-type Htt should therefore be taken into account in the development of a potential therapy, as well as that of mHtt [35].
The structure of Htt in complex with HAP40 has been recently solved by cryo-EM at 4 Å resolution [17]. Conversely, unbound Htt is very difficult to study either by X-ray crystallography or cryo-EM because it has a large size (it contains 3144 residues and weights 348 kDa), is very flexible, and tends to form aggregates. However, about 28% of Htt residues are not visible in the structure, including the first 91 residues, which comprise the 17 glutamine residue-containing polyQ expansion. In agreement with computational predictions, all secondary structure elements belonging to either Htt or HAP40 resolved in the model are α-helices, most of which (72%) are arranged in HEAT or other tandem repeats.
The cryo-EM structure revealed that Htt is formed by three domains: N-terminal domain and C-terminal domain, both of which contain multiple HEAT repeats, joined to a bridge domain (Figure 4).
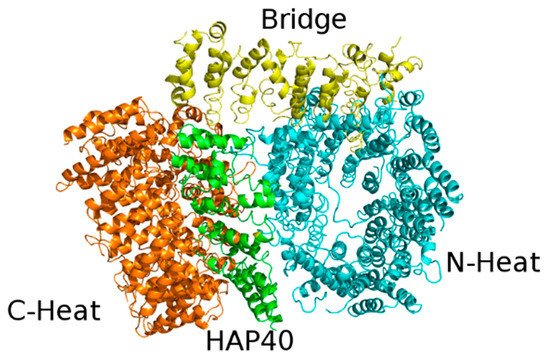
Figure 4. X-ray structure of the complex between HAP40 and Htt (PDB code: 6EZ8). HAP40 is colored green and the C-Heat, Bridge, and N-Heat domains of Htt are colored orange, yellow, and cyan, respectively.
The N-terminal domain (N-HEAT; residues 91–1684) forms a typical α-solenoid, comprising 21 HEAT repeats arranged as a one-and-a-half turn, right-handed superhelix. The N-HEAT was predicted to contain two membrane binding regions: the 1–17 N-terminal tail (not visible in the structure), which was predicted to form an amphipathic helix [36], and a larger region (comprising a.a. 168–366), containing a functionally important palmitoylation site at Cys208 [37]. This region (N-HEAT repeats 2–4) is placed on the N-HEAT convex surface and is positively charged.
The C-HEAT (a.a. 2092–3098) comprises 12 HEAT repeats that form an elliptical ring of ~80 × 30 Å.
The N-HEAT and C-HEAT are joined by the bridge domain. This contains six tandem α-helical repeats, of which repeats three, four and six are Armadillo-like. The repeat region is flanked by five non-repeat helices and a flexible C-terminus (a.a. 2062–2092), which is unresolved.
The lack of interactions between the N-HEAT and C-HEAT domains explains the flexibility of the Htt structure in the absence of interactors. HAP40 binds to a large cleft between these two domains. Within the complex, Htt and HAP40 share large interfaces that comprise mostly hydrophobic interactions. Interestingly, bioinformatic analyses has provided evidence of the evolutionary conservation of these interfaces in all Htt-encoding animal species [38].
Recently, the structure of wild-type Htt in complex with HAP40 has been compared with other mHtt–HAP40 complexes differing in the length of the polyQ repeat, namely, 46QHtt–HAP40 (46 being a typical polyQ length in HD patients) and 128QHtt–HAP40 (128 being an extremely high polyQ length). Quite surprisingly, both crosslinking mass spectrometry and cryo-EM experiments revealed no major structural differences among the different complexes, indicating that the polyQ insertion does not alter the Htt fold [39].
Htt function has not yet been fully elucidated, but Htt has been proposed to be involved in cellular processes such as mitotic spindle orientation, autophagy, and vesicle transport [40][41][42][43]. Htt has been shown to be essential in both embryonal development and adult life. In mice, the Htt knockdown mutant dies at about day 8.5 of gestation [44]. Additionally, Htt deletion in the mouse central nervous system leads to a phenotype similar to that of HD, i.e., cellular stress, neuroinflammation, aberrant synaptic connectivity, and neuronal death [45][46][47].
Htt is crucial for energetic metabolism not only in the brain [48] but also in peripheral tissues. In Htt-null cardiomyocytes, both intracellular ATP and total purine concentration in the cellular medium were reduced. This indicates that, in the heart, Htt plays an important role in both cellular energy balance and nucleotide metabolism [49].
Recent studies carried out using mouse embryonic stem cell demonstrated that Htt is necessary for mitochondrial structure and function from the earliest stages of embryogenesis, providing a molecular explanation for the early embryonic lethality of Htt knockdown [50].
SPN death in HD has mostly been ascribed to toxic ‘gain-of-function’ by mHtt. However, evidence of Htt ‘loss-of-function’ contribution is also available. Indeed, wild-type Htt has been shown to be neuroprotective and, therefore, able to shield neurons against mHtt toxicity [51]. Additionally, Htt deletion in IP-SPN and DP-SPN leads to a phenotype that resembles the key features of HD, supporting the hypothesis that Htt loss-of-function contributes to SPN pathology in HD [52]. Lowering of wild-type Htt expression has also been shown to affect both health and function of primary monocyte-derived macrophages from healthy human subjects, likely by different mechanisms with respect to those associated with mHtt [53].
3.2. Htt Ubiquitination and SUMOylation
Htt is a protein with high ubiquitination potential. It contains 124 lysine residues, many of which are placed on the protein surface, as shown by cryo-EM structural analysis, and may in principle be ubiquitinated or linked to other Ub-like (Ubl) proteins, such as SUMO (Small Ub-like Modifier).
Ubiquitination and/or SUMOylation has been demonstrated for the 30 Htt lysine residues reported in Table 1. Ubiquitination and SUMOylation processes have been shown to compete for lysine residues K6, K9, and K15, all of which are placed in the N-terminal tail that is not visible in the solved cryo-EM structure [54], and they may compete for other lysine residues as well.
Table 1. List of ubiquitinated and SUMOylated residues in human Htt, according to PhosphoSitePlus (phosphosite.org).
| Residue (Human Htt) | Demonstrated Modification | Reference |
|---|---|---|
| Lys 6 | SUMOylation | [54] |
| Lys 9 | SUMOylation | [54] |
| Lys 253 | Ubiquitination | [55] |
| Lys 335 | Ubiquitination | [56] |
| Lys 442 | Ubiquitination | [57] |
| Lys 662 | Ubiquitination | [56] |
| Lys 667 | Ubiquitination | [55] |
| Lys 698 | Ubiquitination | [55] |
| Lys 813 | Ubiquitination | [55] |
| Lys 902 | Ubiquitination | [55] |
| Lys 937 | Ubiquitination | [55] |
| Lys 941 | Ubiquitination | [58] |
| Lys 1121 | Ubiquitination | [56] |
| Lys 1223 | Ubiquitination | [56] |
| Lys 1244 | Ubiquitination | [56] |
| Lys 1262 | Ubiquitination | [59] |
| Lys 1337 | Ubiquitination | [55] |
| Lys 1402 | Ubiquitination | [55][56][58][59][60] |
| Lys 1410 | Ubiquitination | [55][56][58] |
| Lys 1415 | Ubiquitination | [56] |
| Lys 1431 | Ubiquitination | [56][58][61] |
| Lys 1885 | Ubiquitination | [55] |
| Lys 2417 | Ubiquitination | [59] |
| Lys 2423 | Ubiquitination | [56] |
| Lys 2443 | Ubiquitination | [56] |
| Lys 2537 | Ubiquitination | [55] |
| Lys 2564 | Ubiquitination | [55] |
| Lys 2757 | Ubiquitination | [55] |
| Lys 2901 | Ubiquitination | [55] |
| Lys 2967 | Ubiquitination | [55] |
Ubiquitination has been linked to reduced mHtt toxicity, most likely due to increased mHtt clearance by the proteasome [62]. Conversely, SUMOylation has been shown to stabilize mHtt, reduce mHtt aggregation, enhance transcriptional dysregulation by mHtt, and increase mHtt toxicity in a Drosophila model [54]. SUMOylation contribution to mHtt toxicity may be mediated by mHtt targeting of the nucleus and the hampering of ubiquitination and subsequent degradation. While Htt ubiquitination is mediated by different E3 ligases, mHtt SUMOylation is mediated by only one protein, i.e., RHES (Ras homolog enriched in striatum) [63]. RHES has higher affinity for mHtt than wild-type Htt, and its selective expression in the striatum strongly suggests that this protein contributes to the HD pathology [64]. For these reasons, RHES has been considered to be an attractive target for HD therapy [65].
As described above, the presence of seven lysine residues allows Ub to transmit different signals on target proteins. Therefore, Htt polyubiquitination can take place with different mechanisms, which have not yet been completely understood. Two among the 40 E2 enzymes present in the human genome have been demonstrated to interact with Htt: UBE2W, which is known to target the N-termini of disordered proteins such as Tau, RBPB8, and ATXN3, as well as mHtt [66]; and UBE2K, also known as Htt-interacting protein (Hip2) or E2-25k, which is a Ub-conjugating enzyme that directly interacts with Htt and may mediate its ubiquitination [67].
References
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2013, 29, 105–114.
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Epidemiology of Huntington disease. Handb. Clin. Neurol. 2017, 144, 31–46.
- Crowell, V.; Houghton, R.; Tomar, A.; Fernandes, T.; Squitieri, F. Modeling Manifest Huntington’s Disease Prevalence Using Di-agnosed Incidence and Survival Time. Neuroepidemiology 2021, 55, 361–368.
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2147–2157.
- Mielcarek, M.; Smolenski, R.; Isalan, M. Transcriptional Signature of an Altered Purine Metabolism in the Skeletal Muscle of a Huntington’s Disease Mouse Model. Front. Physiol. 2017, 8, 127.
- Mielcarek, M. Huntington’s disease is a multi-system disorder. Rare Dis. 2015, 3, e1058464.
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A Worldwide Study of the Huntington’s Disease Mutation: The Sensitivity and Specificity of Measuring CAG Repeats. N. Engl. J. Med. 1994, 330, 1401–1406.
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993.
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.-J. Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci. 1999, 19, 2522–2534.
- Quarrell, O.; O’Donovan, K.L.; Bandmann, O.; Strong, M. The Prevalence of Juvenile Huntington’s Disease: A Review of the Lit-erature and Meta-Analysis. PLoS Curr. 2012, 4, e4f8606b742ef3.
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 2018, 17, 986–993.
- Tydlacka, S.; Wang, C.-E.; Wang, X.; Li, S.; Li, X.-J. Differential Activities of the Ubiquitin-Proteasome System in Neurons versus Glia May Account for the Preferential Accumulation of Misfolded Proteins in Neurons. J. Neurosci. 2008, 28, 13285–13295.
- Li, X.-J.; Li, H.; Li, S. Clearance of mutant huntingtin. Autophagy 2010, 6, 663–664.
- Li, X.; Wang, C.-E.; Huang, S.; Xu, X.; Li, X.-J.; Li, H.; Li, S. Inhibiting the ubiquitin–proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 2010, 19, 2445–2455.
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Proteostasis Through the Protein Quality Control Function of the Hsp90/Hsp70-based Chaperone Machinery for Treatment of Adult Onset Neurodegenerative Diseases. Annu. Rev. Pharm. Toxicol. 2015, 55, 353–371.
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.-Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254.
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T. Europe PMC Funders Group The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120.
- Qi, L.; Zhang, X.D.; Wu, J.C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.H. The Role of Chaperone-Mediated Autophagy in Huntingtin Deg-radation. PLoS ONE 2012, 7, e46834.
- Baldo, B.; Weiss, A.; Parker, C.N.; Bibel, M.; Paganetti, P.; Kaupmann, K. A Screen for Enhancers of Clearance Identifies Huntingtin as a Heat Shock Protein 90 (Hsp90) Client Protein. J. Biol. Chem. 2012, 287, 1406–1414.
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479.
- Ciechanover, A.; Elias, S.; Heller, H.; Hershko, A. "Covalent affinity" purification of ubiquitin-activating enzyme. J. Biol. Chem. 1982, 257, 2537–2542.
- Hershko, A.; Heller, H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem. Biophys. Res. Commun. 1985, 128, 1079–1086.
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229.
- Ye, Y.; Blaser, G.; Horrocks, M.H.; Ruedas-Rama, M.J.; Ibrahim, S.; Zhukov, A.A.; Orte, A.; Klenerman, D.; Jackson, S.E.; Komander, D. Ubiquitin chain conformation regulates recog-nition and activity of interacting proteins. Nature 2012, 492, 266–270.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Grice, G.L.; Lobb, I.; Weekes, M.; Gygi, S.P.; Antrobus, R.; Nathan, J.A. The Proteasome Distinguishes between Heterotypic and Homotypic Lysine-11-Linked Polyubiquitin Chains. Cell Rep. 2015, 12, 545–553.
- Dougherty, S.E.; Maduka, A.O.; Inada, T.; Silva, G.M. Expanding Role of Ubiquitin in Translational Control. Int. J. Mol. Sci. 2020, 21, 1151.
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133.
- Bremm, A.; Freund, S.M.V.; Komander, D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol. 2010, 17, 939–947.
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209.
- Min, M.; Mevissen, T.E.T.; De Luca, M.; Komander, D.; Lindon, C. Effcient APC/C substrate degradation in cells undergoing mitotic exit depends on K11 ubiquitin linkages. Mol. Biol. Cell 2015, 26, 4325–4332.
- Saito, K.; Horikawa, W.; Ito, K. Inhibiting K63 Polyubiquitination Abolishes No-Go Type Stalled Translation Surveillance in Saccharomyces cerevisiae. PLoS Genet. 2015, 11, e1005197.
- Silva, G.M.; Finley, D.; Vogel, C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct. Mol. Biol. 2015, 22, 116–123.
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P.J. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577.
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78.
- Arndt, J.R.; Chaibva, M.; Legleiter, J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomol. Concepts 2015, 6, 33–46.
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.; et al. Palmitoylation of huntingtin by HIP14is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831.
- Seefelder, M.; Alva, V.; Huang, B.; Engler, T.; Baumeister, W.; Guo, Q.; Fernandez-Busnadiego, R.; Lupas, A.N.; Kochanek, S. The evolution of the huntingtin-associated protein 40 (HAP40) in conjunction with huntingtin. BMC Evol. Biol. 2020, 20, 162.
- Huang, B.; Guo, Q.; Niedermeier, M.L.; Cheng, J.; Engler, T.; Maurer, M.; Pautsch, A.; Baumeister, W.; Stengel, F.; Kochanek, S.; et al. Pathological polyQ expansion does not alter the con-formation of the Huntingtin-HAP40 complex. Structure 2021, 29, 804–809.
- Rui, Y.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature 2015, 17, 262–275.
- Caviston, J.P.; Ross, J.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050.
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138.
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.-L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin Is Required for Mitotic Spindle Orientation and Mammalian Neurogenesis. Neuron 2010, 67, 392–406.
- Zeitlin, S.; Liu, J.-P.; Chapman, D.; Papaioannou, V.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163.
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin Is Required for Normal Excitatory Synapse Development in Cortical and Striatal Circuits. J. Neurosci. 2014, 34, 9455–9472.
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000, 26, 300–306.
- Mehler, M.F.; Petronglo, J.R.; Arteaga-Bracho, E.E.; Gulinello, M.E.; Winchester, M.L.; Pichamoorthy, N.; Young, S.K.; DeJesus, C.D.; Ishtiaq, H.; Gokhan, S.; et al. Loss-of-Huntingtin in Medial and Lateral Ganglionic Lineages Differentially Disrupts Regional Interneuron and Projection Neuron Subtypes and Promotes Huntington’s Disease-Associated Behavioral, Cellular, and Pathological Hallmarks. J. Neurosci. 2019, 39, 1892–1909.
- Morea, V.; Bidollari, E.; Colotti, G.; Fiorillo, A.; Rosati, J.; De Filippis, L.; Squitieri, F.; Ilari, A. Glucose transportation in the brain and its impairment in Huntington disease: One more shade of the energetic metabolism failure? Amino Acids 2017, 49, 1147–1157.
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2020, 1–8.
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin protein is essential for mitochondrial metab-olism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014, 391, 230–240.
- Leavitt, B.R.; van Raamsdonk, J.M.; Shehadeh, J.; Fernandes, H.; Murphy, Z.; Graham, R.K.; Wellington, C.L.; Hayden, M.R. Wild-type huntingtin protects neurons from excitotoxicity. J. Neurochem. 2006, 96, 1121–1129.
- Burrus, C.J.; McKinstry, S.U.; Kim, N.; Ozlu, M.I.; Santoki, A.; Fang, F.Y.; Ma, A.; Karadeniz, Y.B.; Worthington, A.K.; Dragatsis, I.; et al. Striatal Projection Neurons Require Huntingtin for Synaptic Connectivity and Survival. Cell Rep. 2020, 30, 642–657.e6.
- O’Regan, G.C.; Farag, S.H.; Ostroff, G.R.; Tabrizi, S.J.; Andre, R. Wild-type huntingtin regulates human macrophage function. Sci. Rep. 2020, 10, 1–12.
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.-Z.; Cattaneo, E.; et al. SUMO Modification of Huntingtin and Hun-tington’s Disease Pathology. Science 2004, 304, 100–104.
- Kalchman, M.A.; Graham, R.K.; Xia, G.; Koide, H.B.; Hodgson, J.G.; Graham, K.C.; Goldberg, Y.P.; Gietz, R.D.; Pickart, C.M.; Hayden, M.R. Huntingtin Is Ubiquitinated and Interacts with a Specific Ubiquitin-conjugating Enzyme. J. Biol. Chem. 1996, 271, 19385–19394.
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 2009, 324, 1327–1330.
- Falk, J.D.; Vargiu, P.; Foye, P.E.; Usui, H.; Perez, J.; Danielson, P.E.; Lerner, D.L.; Bernal, J.; Sutcliffe, J.G. Rhes: A striatal-specific Ras homolog related to Dexras. J. Neurosci. Res. 1999, 57, 782–788.
- Carbo, M.; Brandi, V.; Pascarella, G.; Staid, D.S.; Colotti, G.; Polticelli, F.; Ilari, A.; Morea, V. Bioinformatics analysis of Ras homologue enriched in the striatum, a potential target for Huntington’s disease therapy. Int. J. Mol. Med. 2019, 44, 2223–2233.
- Wang, B.; Zeng, L.; Merillat, S.A.; Fischer, S.; Ochaba, J.; Thompson, L.M.; Barmada, S.J.; Scaglione, K.M.; Paulson, H.L. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol. Dis. 2017, 109, 127–136.
- De Pril, R.; Fischer, D.F.; Roos, R.A.C.; Van Leeuwen, F.W. Ubiquitin-conjugating enzyme E2-25K increases aggregate formation and cell death in polyglutamine diseases. Mol. Cell Neurosci. 2007, 34, 10–19.
- Maheshwari, M.; Shekhar, S.; Singh, B.K.; Jamal, I.; Vatsa, N.; Kumar, V.; Sharma, A.; Jana, N.R.; Kumar, S.S. Deficiency of Ube3a in Huntington’s disease mice brain increases aggregate load and accelerates disease pathology. Hum. Mol. Genet. 2014, 23, 6235–6245.
- Mishra, A.; Dikshit, P.; Purkayastha, S.; Sharma, J.; Nukina, N.; Jana, N. E6-AP Promotes Misfolded Polyglutamine Proteins for Proteasomal Degradation and Suppresses Polyglutamine Protein Aggregation and Toxicity. J. Biol. Chem. 2008, 283, 7648–7656.
- Joshi, V.; Amanullah, A.; Upadhyay, A.; Mishra, R.; Kumar, A.; Mishra, A. A decade of boon or burden: What has the chip ever done for cellular protein quality control mechanism implicated in neurodegeneration and aging? Front. Mol. Neurosci. 2016, 9, 93.
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase Hrd1 enhances the degradation and sup-presses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550.
- Rubio, I.; Rodríguez-Navarro, J.A.; Tomás-Zapico, C.; Ruíz, C.; Casarejos, M.J.; Perucho, J.; Gomez, A.; Rodal, I.; Lucas, J.J.; Mena, M.A.; et al. Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 2009, 1281, 91–100.
- Bhutani, S.; Das, A.; Maheshwari, M.; Lakhotia, S.; Jana, N. Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 2012, 3, e428.
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzler, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Neurodegeneration
Revisions:
3 times
(View History)
Update Date:
27 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No