 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yanzhuang Wang | + 2290 word(s) | 2290 | 2022-01-17 04:46:54 | | | |
| 2 | Amina Yu | + 541 word(s) | 2831 | 2022-01-19 02:44:19 | | |
Video Upload Options
The Golgi apparatus is an organelle found in most eukaryotic cells. Being part of the endomembrane system in the cytoplasm, it resides at the intersection of the exocytic and endocytic pathways, and works mainly in post-translational modifications and sorting of lipids and proteins. One unique characteristic of the Golgi is the multilayer stack that divides the Golgi membrane system into several sub-compartments known as cis-, medial, and trans-Golgi, each of which contains a set of glycosylation enzymes that sequentially remove or add various sugar monomers to proteins as they pass through the Golgi. To fulfill its function, the Golgi structure is highly dynamic, while Golgi structure and function are tightly regulated. Similarly, the microenvironment of each sub-compartment is also under strict regulation in response to intracellular environmental changes.
1. Ca2+/Mn2+ Transporters and Channels
2. Ca2+/Mn2+ Transporters in the Golgi
3. Ca2+-Release Channels in the Golgi
4. Ca2+-Binding Proteins in the Golgi Lumen
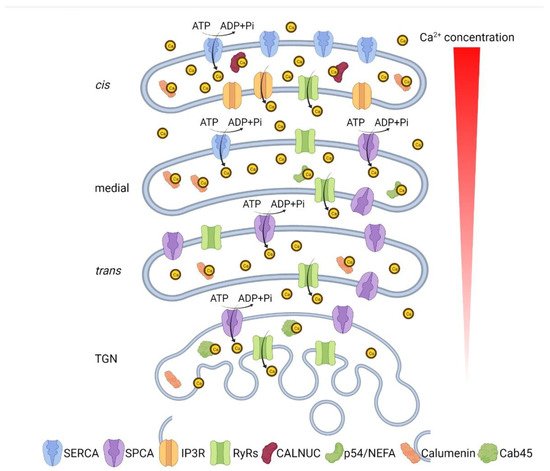
5. Disruption of Ca2+/Mn2+ Homeostasis Impairs Golgi Structure and Function
| Metal Ion | Disease | OMIM | Gene | Protein | Clinical Features |
|---|---|---|---|---|---|
| Ca2+/Mn2+ | Brody myopathy (BRM) [57] | 601003 | ATP2A1 | SERCA1 | Early onset of muscle function disorder characterized by muscle cramping and post-exercise stiffening (myopathy). |
| Acrokeratosis verruciformis (AKV) [58] | 101900 | ATP2A2 | SERCA2 | Early onset keratinization disorder affecting the distal extremities. | |
| Darier disease (DD) [59] | 124200 | ATP2A2 | SERCA2 | Early onset keratinizating disorder characterized by small papules predominantly in seborrheic areas. | |
| Hailey-Hailey disease (HHD) [60] | 169600 | ATP2C1 | SPCA1 | A skin disease causing persistent blisters and suprabasal cell separation (acantholysis) of the epidermis. | |
| Spinocerebellar ataxia 15 (SCA15) [61] | 606658 | ITPR1 | IP3R 1 | A neurological condition characterized by progressive gait and limb ataxia. | |
| Spinocerebellar ataxia 29 (SCA29) [62] | 117360 | ITPR1 | IP3R 1 | Early onset cerebellar ataxia causing slowly progressive or non-progressive gait and limb ataxia. | |
| Gillespie syndrome (GLSP) [15] | 206700 | ITPR1 | IP3R 1 | A congenital neurological disorder characterized by the association of partial bilateral aniridia with non-progressive cerebellar ataxia, and intellectual disability. | |
| Anhidrosis, isolated, with normal sweat glands (ANHD) [63] | 106190 | ITPR2 | IP3R 2 | A disorder characterized by absence of perspiration and subsequent heat intolerance with normal morphology and number of sweat glands. | |
| Malignant hyperthermia 1 (MHS1) [64] | 145600 | RYR1 | RyR1 | A skeletal muscle disorder and the main causes of death due to anesthesia characterized by any combination of hyperthermia, skeletal muscle rigidity, tachycardia or arrhythmia, respiratory and metabolic acidosis, and rhabdomyolysis. | |
| Central core disease of muscle (CCD) [65][66] | 117000 | RYR1 | RyR1 | A mild congenital myopathy characterized by motor developmental delay and signs of mild proximal weakness. | |
| Multiminicore disease with external ophthalmoplegia (MMDO) [67] | 255320 | RYR1 | RyR1 | A heterogeneous neuromuscular disorder characterized by neonatal hypotonia, delayed motor development, and generalized muscle weakness and amyotrophy. | |
| Arrhythmogenic right ventricular dysplasia, familial, 2 (ARVD2) [68] | 600996 | RYR2 | RyR2 | A congenital heart disease characterized by effort-induced polymorphic ventricular tachycardias due to large areas of fatty-fibrous replacement in the subepicardial layer of the right ventricle. | |
| Ventricular tachycardia, catecholaminergic polymorphic, 1 (CPVT1) [69] | 604772 | RYR2 | RyR2 | An arrhythmogenic disorder characterized by physical activity- or stress-induced, polymorphic ventricular tachycardia that may degenerate into deteriorate into ventricular fibrillation. | |
| Congenital disorders of glycosylation, Type IIk (CDG2K) [8] | 614727 | TMEM165 | TMEM165 | An autosomal recessive disorder with a variable phenotype, characterized by growth retardation. | |
| Zn2+ | A novel syndrome with early onset agammaglobulinemia and absent B cells of unknown cause [70] | N/A | SLC39A7 | Zip7 | A novel autosomal recessive disease characterized by absent B cells, agammaglobulinemia and early-onset infections. |
| Ehlers–Danlos syndrome, Spndylodysplastic Type, 3 (SCD-EDS) [71] | 612350 | SLC39A13 | Zip13 | Postnatal growth retardation characterized by short stature, hyperelastic skin and hypermobile joints, protuberant eyes with bluish sclerae, atrophy of the thenar muscles, wrinkled palms and tapering fingers. | |
| Cu2+ | Menkes disease (MNK) [72][73][74] | 309400 | ATP7A | ATP7A | A disorder characterized by generalized copper deficiency, early retardation in growth, peculiar hair, and focal cerebral and cerebellar degeneration due to the dysfunction of several copper-dependent enzymes. |
| Occipital horn syndrome (OHS) [75] | 304150 | ATP7A | ATP7A | A rare connective tissue disorder characterized by hyperelastic and bruisable skin, hernias, bladder diverticula, hyperextensible joints, varicosities, and multiple skeletal abnormalities, sometimes accompanied by mild neurologic impairment, and bony abnormalities of the occiput. | |
| Distal spinal muscular atrophy, X-linked, 3 (DSMAX3) [76] | 300489 | ATP7A | ATP7A | Neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord. | |
| Wilson disease (WD) [77][78] | 277900 | ATP7B | ATP7B | A disorder characterized by dramatic accumulation of intracellular copper with subsequent hepatic and neurologic abnormalities |
6. Golgi Lumenal Ca2+ Is Essential for Intra-Golgi Trafficking and Protein Sorting at the TGN
7. Golgi Ca2+/Mn2+ Homeostasis and Human Diseases
References
- Chandra, S.; Kable, E.P.; Morrison, G.H.; Webb, W.W. Calcium sequestration in the Golgi apparatus of cultured mammalian cells revealed by laser scanning confocal microscopy and ion microscopy. J. Cell Sci. 1991, 100 Pt 4, 747–752.
- Pinton, P.; Pozzan, T.; Rizzuto, R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998, 17, 5298–5308.
- Pizzo, P.; Lissandron, V.; Capitanio, P.; Pozzan, T. Ca2+ signalling in the Golgi apparatus. Cell Calcium 2011, 50, 184–192.
- Vanoevelen, J.; Raeymaekers, L.; Parys, J.B.; De Smedt, H.; Van Baelen, K.; Callewaert, G.; Wuytack, F.; Missiaen, L. Inositol trisphosphate producing agonists do not mobilize the thapsigargin-insensitive part of the endoplasmic-reticulum and Golgi Ca2+ store. Cell Calcium 2004, 35, 115–121.
- Aulestia, F.J.; Alonso, M.T.; Garcia-Sancho, J. Differential calcium handling by the cis and trans regions of the Golgi apparatus. Biochem. J. 2015, 466, 455–465.
- Behne, M.J.; Tu, C.L.; Aronchik, I.; Epstein, E.; Bench, G.; Bikle, D.D.; Pozzan, T.; Mauro, T.M. Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. J. Investig. Dermatol. 2003, 121, 688–694.
- Micaroni, M.; Perinetti, G.; Berrie, C.P.; Mironov, A.A. The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic 2010, 11, 1315–1333.
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2012, 91, 15–26.
- Rosnoblet, C.; Legrand, D.; Demaegd, D.; Hacine-Gherbi, H.; de Bettignies, G.; Bammens, R.; Borrego, C.; Duvet, S.; Morsomme, P.; Matthijs, G.; et al. Impact of disease-causing mutations on TMEM165 subcellular localization, a recently identified protein involved in CDG-II. Hum. Mol. Genet. 2013, 22, 2914–2928.
- Demaegd, D.; Foulquier, F.; Colinet, A.S.; Gremillon, L.; Legrand, D.; Mariot, P.; Peiter, E.; Van Schaftingen, E.; Matthijs, G.; Morsomme, P. Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6859–6864.
- Potelle, S.; Morelle, W.; Dulary, E.; Duvet, S.; Vicogne, D.; Spriet, C.; Krzewinski-Recchi, M.A.; Morsomme, P.; Jaeken, J.; Matthijs, G.; et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum. Mol. Genet. 2016, 25, 1489–1500.
- Potelle, S.; Dulary, E.; Climer, L.; Duvet, S.; Morelle, W.; Vicogne, D.; Lebredonchel, E.; Houdou, M.; Spriet, C.; Krzewinski-Recchi, M.A.; et al. Manganese-induced turnover of TMEM165. Biochem. J. 2017, 474, 1481–1493.
- Lebredonchel, E.; Houdou, M.; Hoffmann, H.H.; Kondratska, K.; Krzewinski, M.A.; Vicogne, D.; Rice, C.M.; Klein, A.; Foulquier, F. Investigating the functional link between TMEM165 and SPCA1. Biochem. J. 2019, 476, 3281–3293.
- Lin, P.; Yao, Y.; Hofmeister, R.; Tsien, R.Y.; Farquhar, M.G. Overexpression of CALNUC (nucleobindin) increases agonist and thapsigargin releasable Ca2+ storage in the Golgi. J. Cell. Biol. 1999, 145, 279–289.
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Bremond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980.
- Schwarzmann, N.; Kunerth, S.; Weber, K.; Mayr, G.W.; Guse, A.H. Knock-down of the type 3 ryanodine receptor impairs sustained Ca2+ signaling via the T cell receptor/CD3 complex. J. Biol. Chem. 2002, 277, 50636–50642.
- Surroca, A.; Wolff, D. Inositol 1,4,5-trisphosphate but not ryanodine-receptor agonists induces calcium release from rat liver Golgi apparatus membrane vesicles. J. Membr. Biol. 2000, 177, 243–249.
- Cifuentes, F.; Gonzalez, C.E.; Fiordelisio, T.; Guerrero, G.; Lai, F.A.; Hernandez-Cruz, A. A ryanodine fluorescent derivative reveals the presence of high-affinity ryanodine binding sites in the Golgi complex of rat sympathetic neurons, with possible functional roles in intracellular Ca2+ signaling. Cell Signal 2001, 13, 353–362.
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203.
- Gallegos-Gomez, M.L.; Greotti, E.; Lopez-Mendez, M.C.; Sanchez-Vazquez, V.H.; Arias, J.M.; Guerrero-Hernandez, A. The Trans Golgi Region is a Labile Intracellular Ca2+ Store Sensitive to Emetine. Sci. Rep. 2018, 8, 17143.
- Lin, P.; Le-Niculescu, H.; Hofmeister, R.; McCaffery, J.M.; Jin, M.; Hennemann, H.; McQuistan, T.; De Vries, L.; Farquhar, M.G. The mammalian calcium-binding protein, nucleobindin (CALNUC), is a Golgi resident protein. J. Cell. Biol. 1998, 141, 1515–1527.
- Scherer, P.E.; Lederkremer, G.Z.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. Cab45, a novel (Ca2+)-binding protein localized to the Golgi lumen. J. Cell. Biol. 1996, 133, 257–268.
- Von Blume, J.; Alleaume, A.M.; Kienzle, C.; Carreras-Sureda, A.; Valverde, M.; Malhotra, V. Cab45 is required for Ca2+-dependent secretory cargo sorting at the trans-Golgi network. J. Cell. Biol. 2012, 199, 1057–1066.
- Hecht, T.K.; Blank, B.; Steger, M.; Lopez, V.; Beck, G.; Ramazanov, B.; Mann, M.; Tagliabracci, V.; von Blume, J. Fam20C regulates protein secretion by Cab45 phosphorylation. J. Cell. Biol. 2020, 219, e201910089.
- Crevenna, A.H.; Blank, B.; Maiser, A.; Emin, D.; Prescher, J.; Beck, G.; Kienzle, C.; Bartnik, K.; Habermann, B.; Pakdel, M.; et al. Secretory cargo sorting by Ca2+-dependent Cab45 oligomerization at the trans-Golgi network. J. Cell. Biol. 2016, 213, 305–314.
- Morel-Huaux, V.M.; Pypaert, M.; Wouters, S.; Tartakoff, A.M.; Jurgan, U.; Gevaert, K.; Courtoy, P.J. The calcium-binding protein p54/NEFA is a novel luminal resident of medial Golgi cisternae that traffics independently of mannosidase II. Eur. J. Cell. Biol. 2002, 81, 87–100.
- Karabinos, A.; Bhattacharya, D.; Morys-Wortmann, C.; Kroll, K.; Hirschfeld, G.; Kratzin, H.D.; Barnikol-Watanabe, S.; Hilschmann, N. The divergent domains of the NEFA and nucleobindin proteins are derived from an EF-hand ancestor. Mol. Biol. Evol. 1996, 13, 990–998.
- Yabe, D.; Nakamura, T.; Kanazawa, N.; Tashiro, K.; Honjo, T. Calumenin, a Ca2+-binding protein retained in the endoplasmic reticulum with a novel carboxyl-terminal sequence, HDEF. J. Biol. Chem. 1997, 272, 18232–18239.
- Vorum, H.; Hager, H.; Christensen, B.M.; Nielsen, S.; Honore, B. Human calumenin localizes to the secretory pathway and is secreted to the medium. Exp. Cell Res. 1999, 248, 473–481.
- Jung, D.H.; Mo, S.H.; Kim, D.H. Calumenin, a multiple EF-hands Ca2+-binding protein, interacts with ryanodine receptor-1 in rabbit skeletal sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 2006, 343, 34–42.
- Sahoo, S.K.; Kim, D.H. Calumenin interacts with SERCA2 in rat cardiac sarcoplasmic reticulum. Mol. Cells 2008, 26, 265–269.
- Sahoo, S.K.; Kim, T.; Kang, G.B.; Lee, J.G.; Eom, S.H.; Kim, D.H. Characterization of calumenin-SERCA2 interaction in mouse cardiac sarcoplasmic reticulum. J. Biol. Chem. 2009, 284, 31109–31121.
- Ireland, S.; Ramnarayanan, S.; Fu, M.; Zhang, X.; Zhang, J.; Li, J.; Emebo, D.; Wang, Y. Cytosolic Ca2+ Modulates Golgi Structure Through PKCalpha-Mediated GRASP55 Phosphorylation. iScience 2020, 23, 100952.
- Zhang, X.; Wang, Y. GRASPs in Golgi Structure and Function. Front. Cell Dev. Biol. 2015, 3, 84.
- Zhang, X.; Wang, Y. Nonredundant Roles of GRASP55 and GRASP65 in the Golgi Apparatus and Beyond. Trends Biochem. Sci. 2020, 45, 1065–1079.
- Dode, L.; Andersen, J.P.; Raeymaekers, L.; Missiaen, L.; Vilsen, B.; Wuytack, F. Functional comparison between secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and sarcoplasmic reticulum Ca2+-ATPase (SERCA) 1 isoforms by steady-state and transient kinetic analyses. J. Biol. Chem. 2005, 280, 39124–39134.
- Tempel, W.; Karaveg, K.; Liu, Z.J.; Rose, J.; Wang, B.C.; Moremen, K.W. Structure of mouse Golgi alpha-mannosidase IA reveals the molecular basis for substrate specificity among class 1 (family 47 glycosylhydrolase) alpha1,2-mannosidases. J. Biol. Chem. 2004, 279, 29774–29786.
- Anderson, E.D.; VanSlyke, J.K.; Thulin, C.D.; Jean, F.; Thomas, G. Activation of the furin endoprotease is a multiple-step process: Requirements for acidification and internal propeptide cleavage. EMBO J. 1997, 16, 1508–1518.
- Schutzbach, J.S.; Forsee, W.T. Calcium ion activation of rabbit liver alpha 1,2-mannosidase. J. Biol. Chem. 1990, 265, 2546–2549.
- Nishikawa, Y.; Pegg, W.; Paulsen, H.; Schachter, H. Control of glycoprotein synthesis. Purification and characterization of rabbit liver UDP-N-acetylglucosamine:alpha-3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I. J. Biol. Chem. 1988, 263, 8270–8281.
- Fritz, T.A.; Hurley, J.H.; Trinh, L.B.; Shiloach, J.; Tabak, L.A. The beginnings of mucin biosynthesis: The crystal structure of UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-T1. Proc. Natl. Acad. Sci. USA 2004, 101, 15307–15312.
- Wiggins, C.A.; Munro, S. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl. Acad. Sci. USA 1998, 95, 7945–7950.
- Palma, A.S.; Morais, V.A.; Coelho, A.V.; Costa, J. Effect of the manganese ion on human alpha3/4 fucosyltransferase III activity. Biometals 2004, 17, 35–43.
- Brito, C.; Kandzia, S.; Graca, T.; Conradt, H.S.; Costa, J. Human fucosyltransferase IX: Specificity towards N-linked glycoproteins and relevance of the cytoplasmic domain in intra-Golgi localization. Biochimie 2008, 90, 1279–1290.
- Missiaen, L.; Raeymaekers, L.; Dode, L.; Vanoevelen, J.; Van Baelen, K.; Parys, J.B.; Callewaert, G.; De Smedt, H.; Segaert, S.; Wuytack, F. SPCA1 pumps and Hailey-Hailey disease. Biochem. Biophys. Res. Commun. 2004, 322, 1204–1213.
- Olanow, C.W. Manganese-induced parkinsonism and Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 209–223.
- Kaufman, R.J.; Swaroop, M.; Murtha-Riel, P. Depletion of manganese within the secretory pathway inhibits O-linked glycosylation in mammalian cells. Biochemistry 1994, 33, 9813–9819.
- Vanoevelen, J.; Dode, L.; Van Baelen, K.; Fairclough, R.J.; Missiaen, L.; Raeymaekers, L.; Wuytack, F. The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J. Biol. Chem. 2005, 280, 22800–22808.
- Mukhopadhyay, S.; Linstedt, A.D. Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc. Natl. Acad. Sci. USA 2011, 108, 858–863.
- Mukhopadhyay, S.; Linstedt, A.D. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 2012, 335, 332–335.
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1994, 1, 59–64.
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40.
- Forrester, A.; Rathjen, S.J.; Daniela Garcia-Castillo, M.; Bachert, C.; Couhert, A.; Tepshi, L.; Pichard, S.; Martinez, J.; Munier, M.; Sierocki, R.; et al. Functional dissection of the retrograde Shiga toxin trafficking inhibitor Retro-2. Nat. Chem. Biol. 2020, 16, 327–336.
- Tewari, R.; Jarvela, T.; Linstedt, A.D. Manganese induces oligomerization to promote down-regulation of the intracellular trafficking receptor used by Shiga toxin. Mol. Biol. Cell 2014, 25, 3049–3058.
- Tewari, R.; Bachert, C.; Linstedt, A.D. Induced oligomerization targets Golgi proteins for degradation in lysosomes. Mol. Biol. Cell. 2015, 26, 4427–4437.
- Mukhopadhyay, S.; Bachert, C.; Smith, D.R.; Linstedt, A.D. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol. Biol. Cell 2010, 21, 1282–1292.
- Odermatt, A.; Barton, K.; Khanna, V.K.; Mathieu, J.; Escolar, D.; Kuntzer, T.; Karpati, G.; MacLennan, D.H. The mutation of Pro789 to Leu reduces the activity of the fast-twitch skeletal muscle sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA1) and is associated with Brody disease. Hum. Genet. 2000, 106, 482–491.
- Dhitavat, J.; Macfarlane, S.; Dode, L.; Leslie, N.; Sakuntabhai, A.; MacSween, R.; Saihan, E.; Hovnanian, A. Acrokeratosis verruciformis of Hopf is caused by mutation in ATP2A2: Evidence that it is allelic to Darier’s disease. J. Investig. Dermatol. 2003, 120, 229–232.
- Sakuntabhai, A.; Ruiz-Perez, V.; Carter, S.; Jacobsen, N.; Burge, S.; Monk, S.; Smith, M.; Munro, C.S.; O’Donovan, M.; Craddock, N.; et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat. Genet. 1999, 21, 271–277.
- Hu, Z.; Bonifas, J.M.; Beech, J.; Bench, G.; Shigihara, T.; Ogawa, H.; Ikeda, S.; Mauro, T.; Epstein, E.H., Jr. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat. Genet. 2000, 24, 61–65.
- Van de Leemput, J.; Chandran, J.; Knight, M.A.; Holtzclaw, L.A.; Scholz, S.; Cookson, M.R.; Houlden, H.; Gwinn-Hardy, K.; Fung, H.C.; Lin, X.; et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007, 3, e108.
- Huang, L.; Chardon, J.W.; Carter, M.T.; Friend, K.L.; Dudding, T.E.; Schwartzentruber, J.; Zou, R.; Schofield, P.W.; Douglas, S.; Bulman, D.E.; et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 2012, 7, 67.
- Klar, J.; Hisatsune, C.; Baig, S.M.; Tariq, M.; Johansson, A.C.; Rasool, M.; Malik, N.A.; Ameur, A.; Sugiura, K.; Feuk, L.; et al. Abolished InsP3R2 function inhibits sweat secretion in both humans and mice. J. Clin. Investig. 2014, 124, 4773–4780.
- MacLennan, D.H.; Duff, C.; Zorzato, F.; Fujii, J.; Phillips, M.; Korneluk, R.G.; Frodis, W.; Britt, B.A.; Worton, R.G. Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature 1990, 343, 559–561.
- Zhang, Y.; Chen, H.S.; Khanna, V.K.; De Leon, S.; Phillips, M.S.; Schappert, K.; Britt, B.A.; Browell, A.K.; MacLennan, D.H. A mutation in the human ryanodine receptor gene associated with central core disease. Nat. Genet. 1993, 5, 46–50.
- Quane, K.A.; Healy, J.M.; Keating, K.E.; Manning, B.M.; Couch, F.J.; Palmucci, L.M.; Doriguzzi, C.; Fagerlund, T.H.; Berg, K.; Ording, H.; et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 1993, 5, 51–55.
- Monnier, N.; Ferreiro, A.; Marty, I.; Labarre-Vila, A.; Mezin, P.; Lunardi, J. A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum. Mol. Genet. 2003, 12, 1171–1178.
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194.
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200.
- Anzilotti, C.; Swan, D.J.; Boisson, B.; Deobagkar-Lele, M.; Oliveira, C.; Chabosseau, P.; Engelhardt, K.R.; Xu, X.; Chen, R.; Alvarez, L.; et al. An essential role for the Zn(2+) transporter ZIP7 in B cell development. Nat. Immunol. 2019, 20, 350–361.
- Giunta, C.; Elcioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305.
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13.
- Chelly, J.; Tumer, Z.; Tonnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19.
- Mercer, J.F.; Livingston, J.; Hall, B.; Paynter, J.A.; Begy, C.; Chandrasekharappa, S.; Lockhart, P.; Grimes, A.; Bhave, M.; Siemieniak, D.; et al. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993, 3, 20–25.
- Kaler, S.G.; Gallo, L.K.; Proud, V.K.; Percy, A.K.; Mark, Y.; Segal, N.A.; Goldstein, D.S.; Holmes, C.S.; Gahl, W.A. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat. Genet. 1994, 8, 195–202.
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352.
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337.
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350.
- Beckers, C.J.; Balch, W.E. Calcium and GTP: Essential components in vesicular trafficking between the endoplasmic reticulum and Golgi apparatus. J. Cell. Biol. 1989, 108, 1245–1256.
- Colombo, M.I.; Beron, W.; Stahl, P.D. Calmodulin regulates endosome fusion. J. Biol. Chem. 1997, 272, 7707–7712.
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell. Biol. 2000, 149, 1053–1062.
- Di Jeso, B.; Pereira, R.; Consiglio, E.; Formisano, S.; Satrustegui, J.; Sandoval, I.V. Demonstration of a Ca2+ requirement for thyroglobulin dimerization and export to the golgi complex. Eur. J. Biochem. 1998, 252, 583–590.
- Ivessa, N.E.; De Lemos-Chiarandini, C.; Gravotta, D.; Sabatini, D.D.; Kreibich, G. The Brefeldin A-induced retrograde transport from the Golgi apparatus to the endoplasmic reticulum depends on calcium sequestered to intracellular stores. J. Biol. Chem. 1995, 270, 25960–25967.
- Micaroni, M. Calcium around the Golgi apparatus: Implications for intracellular membrane trafficking. Adv. Exp. Med. Biol. 2012, 740, 439–460.
- Porat, A.; Elazar, Z. Regulation of intra-Golgi membrane transport by calcium. J. Biol. Chem. 2000, 275, 29233–29237.
- Huttner, W.B.; Ohashi, M.; Kehlenbach, R.H.; Barr, F.A.; Bauerfeind, R.; Braunling, O.; Corbeil, D.; Hannah, M.; Pasolli, H.A.; Schmidt, A.; et al. Biogenesis of neurosecretory vesicles. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 315–327.
- Von Blume, J.; Alleaume, A.M.; Cantero-Recasens, G.; Curwin, A.; Carreras-Sureda, A.; Zimmermann, T.; van Galen, J.; Wakana, Y.; Valverde, M.A.; Malhotra, V. ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev. Cell 2011, 20, 652–662.
- Okunade, G.W.; Miller, M.L.; Azhar, M.; Andringa, A.; Sanford, L.P.; Doetschman, T.; Prasad, V.; Shull, G.E. Loss of the Atp2c1 secretory pathway Ca2+-ATPase (SPCA1) in mice causes Golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J. Biol. Chem. 2007, 282, 26517–26527.
- Deng, Y.; Pakdel, M.; Blank, B.; Sundberg, E.L.; Burd, C.G.; von Blume, J. Activity of the SPCA1 Calcium Pump Couples Sphingomyelin Synthesis to Sorting of Secretory Proteins in the Trans-Golgi Network. Dev. Cell 2018, 47, 464–478.e8.
- Li, L.H.; Tian, X.R.; Jiang, Z.; Zeng, L.W.; He, W.F.; Hu, Z.P. The Golgi Apparatus: Panel Point of Cytosolic Ca2+ Regulation. Neurosignals 2013, 21, 272–284.
- Hoffmann, H.H.; Schneider, W.M.; Blomen, V.A.; Scull, M.A.; Hovnanian, A.; Brummelkamp, T.R.; Rice, C.M. Diverse Viruses Require the Calcium Transporter SPCA1 for Maturation and Spread. Cell Host Microbe 2017, 22, 460–470.e5.
- Wu, C.; Zheng, M.; Yang, Y.; Gu, X.; Yang, K.; Li, M.; Liu, Y.; Zhang, Q.; Zhang, P.; Wang, Y.; et al. Furin: A Potential Therapeutic Target for COVID-19. iScience 2020, 23, 101642.

