The Golgi apparatus is an organelle found in most eukaryotic cells. Being part of the endomembrane system in the cytoplasm, it resides at the intersection of the exocytic and endocytic pathways, and works mainly in post-translational modifications and sorting of lipids and proteins. One unique characteristic of the Golgi is the multilayer stack that divides the Golgi membrane system into several sub-compartments known as cis-, medial, and trans-Golgi, each of which contains a set of glycosylation enzymes that sequentially remove or add various sugar monomers to proteins as they pass through the Golgi. To fulfill its function, the Golgi structure is highly dynamic, while Golgi structure and function are tightly regulated. Similarly, the microenvironment of each sub-compartment is also under strict regulation in response to intracellular environmental changes.
- Golgi
- homeostasis
- transporter
- channel
- metal ion
- calcium
- manganese
- zinc
- copper
1. Ca2+/Mn2+ Transporters and Channels
2. Ca2+/Mn2+ Transporters in the Golgi
3. Ca2+-Release Channels in the Golgi
4. Ca2+-Binding Proteins in the Golgi Lumen
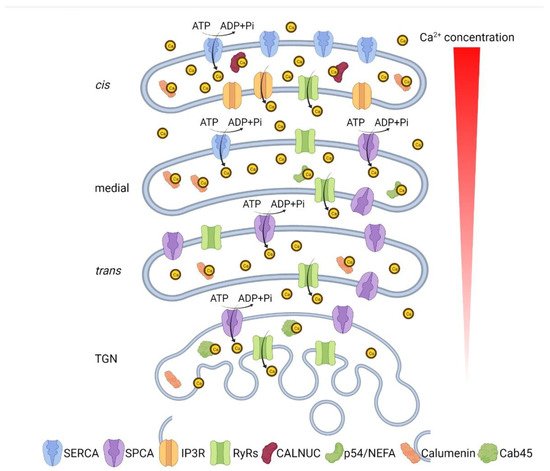
5. Disruption of Ca2+/Mn2+ Homeostasis Impairs Golgi Structure and Function
| Metal Ion | Disease | OMIM | Gene | Protein | Clinical Features |
|---|---|---|---|---|---|
| Ca2+/Mn2+ | Brody myopathy (BRM) [57] | 601003 | ATP2A1 | SERCA1 | Early onset of muscle function disorder characterized by muscle cramping and post-exercise stiffening (myopathy). |
| Acrokeratosis verruciformis (AKV) [58] | 101900 | ATP2A2 | SERCA2 | Early onset keratinization disorder affecting the distal extremities. | |
| Darier disease (DD) [59] | 124200 | ATP2A2 | SERCA2 | Early onset keratinizating disorder characterized by small papules predominantly in seborrheic areas. | |
| Hailey-Hailey disease (HHD) [60] | 169600 | ATP2C1 | SPCA1 | A skin disease causing persistent blisters and suprabasal cell separation (acantholysis) of the epidermis. | |
| Spinocerebellar ataxia 15 (SCA15) [61] | 606658 | ITPR1 | IP3R 1 | A neurological condition characterized by progressive gait and limb ataxia. | |
| Spinocerebellar ataxia 29 (SCA29) [62] | 117360 | ITPR1 | IP3R 1 | Early onset cerebellar ataxia causing slowly progressive or non-progressive gait and limb ataxia. | |
| Gillespie syndrome (GLSP) [15] | 206700 | ITPR1 | IP3R 1 | A congenital neurological disorder characterized by the association of partial bilateral aniridia with non-progressive cerebellar ataxia, and intellectual disability. | |
| Anhidrosis, isolated, with normal sweat glands (ANHD) [63] | 106190 | ITPR2 | IP3R 2 | A disorder characterized by absence of perspiration and subsequent heat intolerance with normal morphology and number of sweat glands. | |
| Malignant hyperthermia 1 (MHS1) [64] | 145600 | RYR1 | RyR1 | A skeletal muscle disorder and the main causes of death due to anesthesia characterized by any combination of hyperthermia, skeletal muscle rigidity, tachycardia or arrhythmia, respiratory and metabolic acidosis, and rhabdomyolysis. | |
| Central core disease of muscle (CCD) [65][66] | 117000 | RYR1 | RyR1 | A mild congenital myopathy characterized by motor developmental delay and signs of mild proximal weakness. | |
| Multiminicore disease with external ophthalmoplegia (MMDO) [67] | 255320 | RYR1 | RyR1 | A heterogeneous neuromuscular disorder characterized by neonatal hypotonia, delayed motor development, and generalized muscle weakness and amyotrophy. | |
| Arrhythmogenic right ventricular dysplasia, familial, 2 (ARVD2) [68] | 600996 | RYR2 | RyR2 | A congenital heart disease characterized by effort-induced polymorphic ventricular tachycardias due to large areas of fatty-fibrous replacement in the subepicardial layer of the right ventricle. | |
| Ventricular tachycardia, catecholaminergic polymorphic, 1 (CPVT1) [69] | 604772 | RYR2 | RyR2 | An arrhythmogenic disorder characterized by physical activity- or stress-induced, polymorphic ventricular tachycardia that may degenerate into deteriorate into ventricular fibrillation. | |
| Congenital disorders of glycosylation, Type IIk (CDG2K) [8] | 614727 | TMEM165 | TMEM165 | An autosomal recessive disorder with a variable phenotype, characterized by growth retardation. | |
| Zn2+ | A novel syndrome with early onset agammaglobulinemia and absent B cells of unknown cause [70] | N/A | SLC39A7 | Zip7 | A novel autosomal recessive disease characterized by absent B cells, agammaglobulinemia and early-onset infections. |
| Ehlers–Danlos syndrome, Spndylodysplastic Type, 3 (SCD-EDS) [71] | 612350 | SLC39A13 | Zip13 | Postnatal growth retardation characterized by short stature, hyperelastic skin and hypermobile joints, protuberant eyes with bluish sclerae, atrophy of the thenar muscles, wrinkled palms and tapering fingers. | |
| Cu2+ | Menkes disease (MNK) [72][73][74] | 309400 | ATP7A | ATP7A | A disorder characterized by generalized copper deficiency, early retardation in growth, peculiar hair, and focal cerebral and cerebellar degeneration due to the dysfunction of several copper-dependent enzymes. |
| Occipital horn syndrome (OHS) [75] | 304150 | ATP7A | ATP7A | A rare connective tissue disorder characterized by hyperelastic and bruisable skin, hernias, bladder diverticula, hyperextensible joints, varicosities, and multiple skeletal abnormalities, sometimes accompanied by mild neurologic impairment, and bony abnormalities of the occiput. | |
| Distal spinal muscular atrophy, X-linked, 3 (DSMAX3) [76] | 300489 | ATP7A | ATP7A | Neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord. | |
| Wilson disease (WD) [77][78] | 277900 | ATP7B | ATP7B | A disorder characterized by dramatic accumulation of intracellular copper with subsequent hepatic and neurologic abnormalities |