Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Genesis Danielle Flores | + 4800 word(s) | 4800 | 2021-12-20 03:47:42 | | | |
| 2 | Bruce Ren | Meta information modification | 4800 | 2022-01-17 02:11:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Flores, G. The Brain-Gut-Microbiome System and Autism Spectrum Disorder. Encyclopedia. Available online: https://encyclopedia.pub/entry/18276 (accessed on 24 July 2026).
Flores G. The Brain-Gut-Microbiome System and Autism Spectrum Disorder. Encyclopedia. Available at: https://encyclopedia.pub/entry/18276. Accessed July 24, 2026.
Flores, Genesis. "The Brain-Gut-Microbiome System and Autism Spectrum Disorder" Encyclopedia, https://encyclopedia.pub/entry/18276 (accessed July 24, 2026).
Flores, G. (2022, January 14). The Brain-Gut-Microbiome System and Autism Spectrum Disorder. In Encyclopedia. https://encyclopedia.pub/entry/18276
Flores, Genesis. "The Brain-Gut-Microbiome System and Autism Spectrum Disorder." Encyclopedia. Web. 14 January, 2022.
Copy Citation
Gastrointestinal dysfunction is one of the most prevalent physiological symptoms of autism spectrum disorder (ASD). A growing body of largely preclinical research suggests that dysbiotic gut microbiota may modulate brain function and social behavior, yet little is known about the mechanisms that underlie these relationships and how they may influence the pathogenesis or severity of ASD.
autism spectrum disorder
brain-gut-microbiome system
gut-brain axis
microbiome
probiotics
tryptophan pathway
1. Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder characterized by two core deficits: persistent difficulties in social communication and interaction, and restricted, repetitive patterns of behavior [1]. A growing body of research suggests that gut microbiota may serve an important role in modulating brain function, social behavior, and ASD symptomatology; for a review, see [2].
2. The Brain-Gut-Microbiome (BGM) System
Over the past decade, a surge of research has emerged that investigates the bidirectional relationship between the brain and the human gut microbiome, known as the brain-gut-microbiome (BGM) system. Gut microbiota play a crucial role in the modulation of cross-talk between the gut and nervous system [3] and promote gastrointestinal (GI) homeostasis, in addition to impacting higher cognitive functions [2]. The BGM system includes the central and enteric nervous systems (CNS; ENS) and various neural, metabolic, endocrine, and immune mediators [2]. Some microbiota, as well as their molecular by-products, neuroactive metabolites, and related inflammatory mediators, can cross both the gut and blood-brain barriers, allowing transmission along the BGM system [4].
The BGM system closely interacts with several other biological systems that regulate the body, including the immune system, hypothalamic-pituitary-adrenal axis, and the two branches of the autonomic nervous system (Figure 1) [5][6]. The afferent vagus nerve transmits information from visceral organs to brain regions, such as the hypothalamus, amygdala, and the insular cortex [7], as well as brainstem nuclei, which in turn are critical for the bidirectional communication between the gut and the brain [8]. These multiple lines of communication work in conjunction, allowing the brain and gut to influence each other [6]. Research on how the BGM system influences cognition and behavior, in particular, has been explored over the last decade; however, it is still largely in its nascent stages.
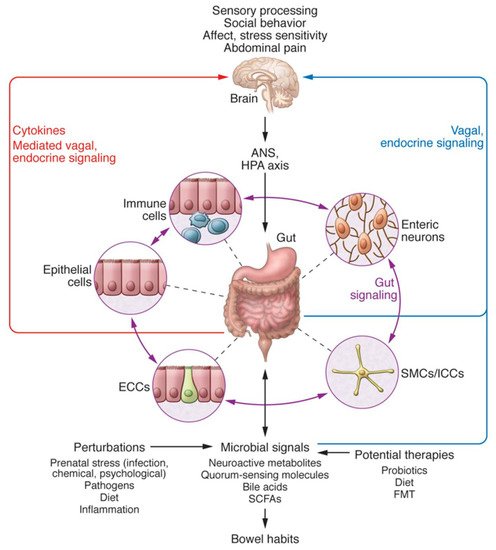
Figure 1. Bidirectional interactions within the Brain-Gut-Microbiome (BGM) System taken from Mayer et al. [6]. The BGM system comprises a complicated network with multiple feedback loops that allow signaling between microbiota and the brain and gut connectomes. The microbiome can modulate sensory processing, social behavior, affect, and the two arms of the stress response, in addition to abdominal pain, directly via various neuroactive and inflammatory signaling molecules, or indirectly via the vagus nerve. In turn, the brain can modulate gut microbial composition and function directly by the release of neuroactive compounds into the gut lumen acting on receptors of certain gut microbes, or via the regulation of intestinal motility and secretion activities, indirectly affecting the composition and functions of the gut microbiome. Both prenatal and postnatal perturbations to the BGM system, including but not limited to diet, infection, inflammation, and psychosocial stress, can influence the stability of these neural, neuroendocrine and immunoregulatory communication channels to create fundamental changes in brain structure and function. ANS = autonomic nervous system; HPA = hypothalamic-pituitary-adrenal; SMC = smooth muscle cells; ICC = interstitial cells of Cajal; ECC = enterochromaffin cells; SCFAs = short-chain fatty acids; FMT = fecal microbial transplant. Copyright © 2021, American Society for Clinical Investigation. The request has been put in and we are waiting for documentation.
2.1. Gut Microbiota and Development
Gut microbiota development during the first 1000 days of life (including prenatal life) is critical for establishing a healthy and protective microbiome (Figure 2) [9]. Data indicate that one’s microbiome begins to develop rapidly following birth, with influencing factors such as the delivery method, infant feeding practices, antibiotics, and the environment [10]. Early life dysbiosis may be especially impactful in early neurodevelopment with the potential to alter the integrity of the blood-brain barrier and alter brain-gut signaling, both of which can lead to adverse health outcomes later in life [11][12]. In addition, prenatal maternal factors, including psychosocial stress, infections, obesity, and metabolic syndrome, can result in maternal dysbiosis and dysregulated maternal immune activation, posing significant risks to offspring neurodevelopment [13]. Throughout early life and childhood, microbiota continue to play major roles in modulating immune system functioning, as well as the maturation of the brain and body of the host [11][14][15][16].
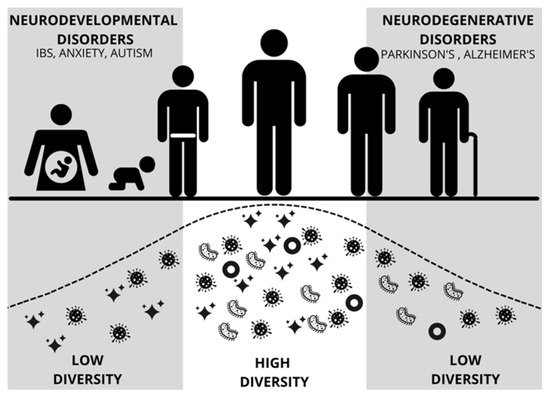
Figure 2. Figure modified from Mayer [9] depicting diversity and abundance of gut microbes across the lifespan of a human. Early and late periods of low diversity coincide with vulnerability to neurodevelopmental disorders and neurodegenerative disorders, respectively. IBS = Irritable Bowel Syndrome.
Similarly, in early neural development, the developing microbiome appears to be particularly sensitive during early life and its profile can be altered by external stimuli, including stress, adversity, diet, environmental microbes, and antibiotics, with both immediate and long-term negative effects on the integrity of the immune system, metabolism, and overall health [17][18][19]. Such perturbations during sensitive periods of development have also been linked to negative cognitive outcomes [20][21][22], socioemotional functioning [23][24][25][26], and internalizing and externalizing problem behaviors [21][22], all of which have serious implications for various neurodevelopmental disorders.
2.2. Gut Microbiota and Clinical Symptomatology in ASD
Previous research has shown an association between gut microbiota and various neuropsychiatric disorders, including attention deficit hyperactivity disorder (ADHD), depression, and obsessive-compulsive disorder [27], which often co-occur with ASD [28]. ASD symptomatology has also been directly associated with gut microbiota [29]. Based on these reported associations and the multitude of potential therapeutic targets, new therapeutic interventions targeting the BGM system have been proposed and evaluated [30][31]. Below, we focus on current research that informs our understanding of gut microbiota and ASD, including GI disorders, social deficits, disruptions in neurochemical mechanisms, and abnormal brain structure and function.
3. Gut Microbiota and ASD Symptomatology
3.1. Gut Microbiota and GI Impairment in ASD
GI symptoms—abdominal pain, constipation, and diarrhea, in particular—have been reported in 46–84% of individuals with ASD [32], which has led to the hypothesis that gut dysbiosis may be especially relevant to ASD patients with GI distress. Studies investigating the gut microbiome of children with ASD have found abnormal gut-derived metabolite patterns as well as certain taxa that significantly differ in relative abundance from healthy controls (e.g., Clostridia, Desulfovibrio, Bifidobacteria, Bacteroides) and are strongly associated with GI symptoms [33][34][35][36][37][38][39]. To date, the exact microbial composition associated with ASD has yet to be determined, with contradictory findings existing at the phylum, genus, and species levels, as well as in alpha and beta diversity; for a review, see [34]. It is important to note that this lack of consensus may be due to several factors, including study-wide differences in collection methods, preprocessing, statistical analysis, age, sex, participant diet, specimen type, ASD heterogeneity, and presence of GI disorders, all of which demonstrate a need for more homogenous samples and standardized collection and analysis procedures [40].
3.2. Gut Microbiota and ASD-Related Behavior
It has been theorized that a long history of coevolution has closely linked social behavior and gut microbiota across the animal kingdom [41]. A bidirectional relationship has been observed in animal models in which the gut microbiome influences social behavior, while social interactions and social structures also shape the composition and function of the microbiome [41][42][43]. Much of the work tying gut microbiota to social behavior has been conducted in rodent models [44], including germ-free rodents [45][46][47] and mouse models known for their phenotypic similarities to humans with ASD (e.g., repetitive movements, low reciprocal social interactions) [48][49]. These studies have utilized both bottom-up (e.g., manipulating the presence of bacteria by colonizing germ-free mice) [47] and top-down (e.g., starting with a genetic mouse model and investigating their gut microbiome) [48] approaches.
In experiments conducted by Desbonnet et al. [47], male mice under germ-free rearing conditions engaged in social avoidant and repetitive behaviors and displayed a lack of interest in social novelty and social motivation, all of which are considered ASD-like behaviors. When the microbiome of a second set of germ-free mice was colonized with fecal bacteria from neurotypical, normally behaving mice, many of these behavioral deficits were reversed, demonstrating the significance of healthy microbiota for typical social functioning in mouse models. These data support the notion that gut bacteria modulate ASD-like symptoms. Using a top-down approach, Golubeva and colleagues [48] investigated the interaction between GI physiology, microbiota composition, and social behavior in BTBR T+Itpr3tf/J (BTBR) mice, which are well-validated models known to exhibit ASD-like behaviors. They found that BTBR mice displayed altered relative abundance levels in 18 out of 44 identified gut bacterial genera, which have been linked to physiological and behavioral impairments. Specifically, the mice showed significantly reduced amounts of Bifidobacteria and Blautia species, both of which play vital roles in optimal GI and metabolic functioning and social interactions within the BTBR mouse strain. This finding is consistent with those of Wang et al. [50], who compared the fecal samples of children with ASD and neurotypical controls and observed reduced abundances of Bifidobacterium in the ASD group. Additional bacterial taxa, including Akkermansia, Bacteroides, Desulfovibrio, and Lactobacillus, were found to have abnormal abundances in the gut of BTBR mice [48]. These taxa have also been associated with ASD symptomatology, including social, repetitive, and anxious behaviors in both animal and human studies [29][35][50][51][52][53].
In both human subjects and mouse models of ASD, gut microbiota-associated metabolites have been linked to ASD symptoms and co-occurring GI abnormalities [33][34][35][36]. Needham et al. [36], however, reported significant differences between the fecal and plasma metabolomes of typically developing (TD) and ASD children regardless of the presence of GI symptoms in the ASD group, demonstrating that microbial abnormalities and their influence on behavior may not be unique to ASD patients with GI dysfunction. Interestingly, metabolite levels were correlated with clinical behaviors, as measured by two clinical ASD assessments (Autism Diagnostic Observation Schedule [ADOS] and Autism Diagnostic Interview-Revised [ADI-R]), demonstrating significant relationships between metabolites, GI function, and behavior [36]. In another notable human-mouse microbiome study, Sharon and colleagues [54] transferred gut microbiota from ASD and TD hosts into germ-free wild-type mice. After the colonization of ASD microbiota, the mice displayed various hallmark ASD-like behaviors, such as increased repetitive behavior and decreased locomotion and communication, as well as different metabolome profiles compared to offspring mice colonized with microbiota from TD controls [54]. In addition to these behaviors, more recent studies have found decreased sociability, decreased sensitivity to social odors, and dysregulated metabolic pathways and metabolites in germ-free mice colonized with ASD bacteria [36][55]. Taken together, the studies above suggest that altered gut microbiota and some of their metabolites influence ASD-like behaviors in rodent models and ASD symptoms in patients.
3.3. Gut Microbiota Therapy and the Reduction of ASD Symptomatology
3.3.1. Probiotic Therapy
Given the prior findings on microbiota and behavioral symptoms in ASD, a number of studies have investigated the use of probiotics (“live microorganisms which when administered in adequate amounts confer a health benefit on the host” [56]) as a potential treatment. Indeed, probiotics can alleviate GI symptoms in both ASD and TD populations [57][58][59][60]. Thus, the question of whether probiotics can also be used to treat behavioral symptoms of ASD has been explored, particularly in rodent models. Hsiao et al. [49] used a maternal immune activation (MIA) mouse model whose offspring display many core phenotypes of ASD (e.g., altered communication, abnormalities in social behavior, stereotyped behaviors) following prenatal maternal treatment with the viral polyinosinic-polycytidylic acid (poly I:C) during specific points of their neurodevelopment [61]. It was observed that MIA offspring displayed ASD-related behavioral abnormalities while also having increased intestinal permeability, microbiome changes, and metabolomic alterations [49]. Oral administration of the gut commensal Bacteroides fragilis corrected gut permeability, improved gut microbiota and blood metabolite profiles, and ameliorated atypical anxiety, communicative (i.e., ultrasonic vocalizations), repetitive, and sensorimotor behavioral symptoms, but not sociability or social preference, in the MIA offspring (Figure 3) [49].
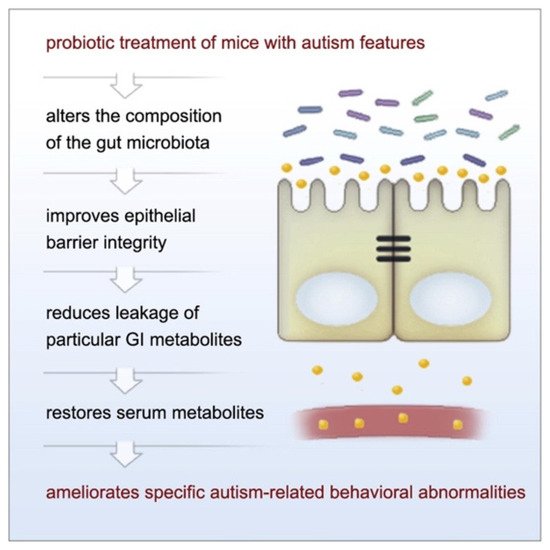
Figure 3. The observed relationship between probiotic use and amelioration of ASD-like symptoms in MIA offspring. Figure taken from Hsiao et al. [49] Graphical Abstract. Cell 2013 155, 1451–1463 DOI: (https://doi.org/10.1016/j.cell.2013.11.024) Copyright © 2021 Elsevier Inc.
Although the amelioration of all social symptoms was not observed in the aforementioned study, preclinical evidence suggests that other strains or types of microbes may improve social behavior [49]. In fact, several studies have demonstrated that various strains of Lactobacillus reduce social impairments in animal models [44]. Buffington et al. [45], for example, found that introducing L. reuteri reduced social deficits in maternal high-fat-diet rodent offspring, which are born with gut-microbial alterations detrimental to their social functioning. In support of using Lactobacillus strains as a means for treating ASD symptoms, a more recent study demonstrated that a L. plantarum PS128 intervention for children with ASD reduced aberrant behaviors commonly seen in individuals with ASD and may ameliorate social communicative impairments [62].
3.3.2. Fecal Microbiota Transplantation Therapy
Fecal microbial transplantation (FMT) has been shown to alleviate the behavioral symptoms of various neuropsychiatric disorders, including ASD [63]. A recent exploratory, unblinded, and non-randomized clinical trial, involving 18 children diagnosed with ASD (with moderate to severe GI issues) and 20 TD children matched by age and gender without GI disorders, evaluated the effect of repeated Microbiota Transfer Therapy (MTT), a modified FMT, on gut microbiota composition and GI and ASD-related symptoms [64]. MTT, which combined antibiotic treatment, a bowel cleanse, a stomach-acid suppressant, and an extended fecal microbiota transplant, led to significant improvements in both GI and ASD symptoms, including improvements in social skill deficits [64]. A majority of these changes were sustained and even improved two years after completion of the treatment [65]. Additionally, it was shown that both the plasma and fecal metabolite profiles of the ASD group became more similar to those of their TD counterparts following MTT [66][67]. The effects of FMT have also been evaluated in adult pathogen-free mice using donor and in vitro-cultured human gut microbiota. Chen et al. [68], for example, reported significant improvements in behavioral impairments associated with ASD, particularly for anxiety-related and repetitive behaviors, as well as moderate improvements in social behavior, with the findings in alignment with previous literature. Taken together, data from rodent models and preliminary clinical studies suggest that interventions involving both probiotics and FMT may offer promising lines of research for understanding ASD and may help in the development of novel therapies.
4. Putative Mechanisms of the BGM System Related to ASD
Several models have been put forth hypothesizing biological mechanisms associated with abnormal gut microbiota and symptoms in ASD. One mechanism relevant to modulating the clinical symptoms of ASD is the metabolism of the essential amino acid tryptophan along the BGM system. Here, we discuss the influence of gut microbiota on the tryptophan metabolic pathways in both animals and humans, as well as how disruptions to these pathways influence ASD social and behavioral deficits (Figure 4).
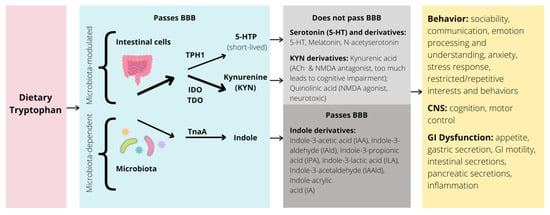
Figure 4. Simplified illustration of the tryptophan pathway in the GI tract and potential impacts on behavior, CNS, and GI dysfunction. BBB = blood brain barrier; TPH = tryptophan hydroxylase; IDO = indolamine 2,3-dioxygenase; TDO = tryptophan 2,3-dioxygenase; TnaA = tryptophanase; 5-HTP = 5-hydroxytryptophan; 5-HT = 5-hydroxytryptamine, or serotonin; ACh = acetylcholine; NMDA = N-methyl-D-aspartate; CNS = central nervous system; GI = gastrointestinal; arrow thickness represents strength of pathway.
4.1. Tryptophan Pathways
Dietary tryptophan is metabolized through three main pathways: the serotonin, kynurenine (KYN), and indole metabolic pathways. Over 95% of tryptophan is oxidized and degraded to yield metabolites along the KYN pathway [69]. Importantly, tryptophan is also the sole precursor to the neurotransmitter serotonin (5-HT) in the brain and gut (synthesized by action of enzyme tryptophan hydroxylase [TPH]) [69]. Whereas gut microbiota play a modulatory role in the balance between serotonin and KYN production, the biosynthesis of indoles and indole derivatives (e.g., indole-3-aldehyde, indole-3-acetic acid, indole-3-propionic acid) from tryptophan is fully dependent on the enzyme tryptophanase, only found in select microbes [70]. How changes in the relative abundance of certain gut microbiota contribute to modifications of these pathways, central tryptophan metabolism, and ultimately brain function and behavior, is a crucial and ongoing area of research [71]. Many of these findings also require analysis of how this relationship modulates clinical symptoms characteristic of neurodevelopmental disorders, such as ASD.
4.1.1. Indole Pathway and ASD
Indole synthesis is driven by certain bacterial taxa that convert undigested tryptophan from the gut lumen into indole and indole derivatives, constituting an exclusively microbe-dependent pathway [70]. Many of the phyla, genera, and species associated with the production of indoles and altered indole products involved in tryptophan metabolism have been linked to the development of ASD and related neuropsychiatric disorders [33][55][72][73][74][75]. Mice from the MIA model have shown abnormally high levels of key serum metabolites produced by gut microbes, including 4-ethylphenylsulfate, serum indolepyruvate, and indole-3-acryloylglycine, all of which were readjusted by treatment with B. fragilis [49]. In comparison, a human study also found that urinary metabolites of ASD and TD children significantly differed along the tryptophan and purine metabolic pathways, suggesting that the gut microbiome contributes to abnormal tryptophan metabolism in ASD [73]. Specifically, gut bacteria-derived metabolites indolyl-3-acetic acid and indolyl-lactate were more numerous in the ASD group compared to controls [73], consistent with the findings from Xiao et al. [55] in the cecal matter of mice that had received FMT from ASD donors. These altered pathways overlapped with those of rodent models which displayed ASD-like behaviors, demonstrating a potential pathophysiological explanation for many behavioral symptoms of ASD [73]. Similarly, De Angelis et al. [72] found increased indole and 3-methylindole in the fecal samples of ASD children. After indole is absorbed in the gut, it is oxidized and sulfated by liver enzymes into indoxyl and indoxyl sulfate metabolites, respectively. Interestingly, these indole metabolites have been identified as potential metabolic markers for ASD, as well [76][77].
4.1.2. Kynurenine Pathway and ASD
The KYN pathway, also derived from tryptophan and modulated by gut microbes, largely depends on indoleamine-2,3-dioxygenase (IDO) and, to a lesser degree, tryptophan-2, 3-dioxygenase (TDO) for metabolization [78]. IDO, expressed in all body tissues, is typically activated in the presence of pro-inflammatory cytokines, whereas TDO, expressed primarily in liver tissues, is activated by glucocorticoids [78][79]. Once transformed from tryptophan, KYN metabolizes into two downstream metabolites, neuroprotective kynurenic acid (KA) and neurotoxic quinolinic acid (QA) [78]. Recent evidence suggests that altered KYN metabolism is indicative of greater tryptophan depletion and an impaired serotonergic pathway in ASD [80]. In a study investigating the role of the KYN pathway in ASD, Bryn et al. [81] showed that children with ASD had significantly lower KA serum levels, higher KYN/KA ratios, and higher QA serum concentrations than TD children. These findings are consistent with those of Gevi et al. [73], who found that tryptophan was disproportionately metabolized into QA, with significantly decreased levels of KA, in children with ASD. Both studies demonstrate an increased potential for neurotoxicity in children with ASD, which is thought to be involved in the pathophysiology of the disorder [73][81]. Interestingly, Xiao et al. [55] found increased KA in mice following FMT from children with ASD. These levels correlated with specific bacteria (e.g., genera in the orders Clostridiales and Bacteroidetes), supporting their modulatory role in tryptophan metabolism, but demonstrating a need for further research on how microbiota alter KYN-pathway products [55].
Although there is minimal literature associating gut microbiota with the KYN pathway in human ASD populations, studies in other clinical and normative populations have provided evidence to support this relationship [78]. Interestingly, Luna et al. [82] found in their study of ASD microbiome-neuroimmune signatures that along with tryptophan and serotonin levels, inflammatory cytokine levels correlated with certain bacterial species in children with ASD and functional GI disorder comorbidities. No direct link was made to the KYN pathway; however, because IDO is typically activated in response to cytokines [78][79], there is reason to investigate whether the abnormal microbial profile of individuals with ASD may be implicated in the dysregulation of the KYN pathway.
4.1.3. Serotonin Pathway and ASD
Serotonin (also referred to as 5-HT) is important for mood regulation, higher order cognition, and neurodevelopment of both the CNS and ENS [83][84]. Although the majority (>90%) of serotonin comes from enterochromaffin cells in the GI tract, serotonin is also synthesized in the neurons of the ENS and CNS, particularly the raphe nuclei in the brainstem [85]. Gut microbiota and their metabolites can influence central and peripheral serotonin production and metabolism through a variety of mechanisms [71][86]. Because only a small percentage of tryptophan is converted into serotonin, any alterations to its metabolism and availability can pose a significant risk to one’s health [87].
Approximately 30% of ASD patients have hyperserotonemia, or elevated whole-blood serotonin levels [88], which is believed to be due in part to increased serotonin production in enterochromaffin cells in the gut [89]. Based on similar and replicated findings, it has been postulated that hyperserotonemia may represent a highly heritable biomarker of ASD and that the serotonin pathway as a whole may be dysfunctional in at least a subgroup of ASD individuals [89][90]. In preclinical models, hyperserotonemia has been linked to social-behavioral deficits characteristic of ASD [49][91][92]. Tanaka et al. [92], for example, found that a tryptophan-depleted diet, which decreases brain serotonin levels and regulates gene expression inside the serotonin system, improved social impairments of genetically modified ASD mouse models. Lim et al.’s [91] report of elevated serum serotonin levels in environmental risk factor mouse models of ASD that were associated with changes in bacteria known to stimulate serotonin production suggests that alterations in serotonin and hyperserotonemia itself may have a microbial origin. The connection between serotonin and the microbiome has been made in humans as well, as demonstrated by a link between increased GI symptom severity and hyperserotonemia in ASD youth [93]. Other studies investigating serotonin-related dysfunction in children with ASD and co-occurring GI symptoms have implicated fecal metabolites in the metabolic network of various neurotransmitters, including serotonin [33], and have found increased levels of serotonergic metabolites, including 5-HIAA, the main metabolite of serotonin, in the rectal tissue of ASD youth with co-occurring functional GI disorders [82]. These metabolite levels correlated with the dysbiosis of several bacterial species, demonstrating a potential microbiome profile for ASD [82].
SERT Ala56, the most common variant of the serotonin-selective transporter responsible for serotonin reuptake in both the brain and intestines, has been found to be overexpressed in ASD patients and linked to neurobiological and GI symptoms in a genetically modified murine ASD model [94]. SERT Ala56 mice are also known to exhibit serotonin-related dysfunction, including excess clearance of central serotonin, augmented serotonin receptor sensitivity, and hyperserotonemia [95]. Research supporting connections between an altered serotonin system and ASD pathophysiology has demonstrated a positive relationship of serotonin and SERT levels with autism symptom severity in humans [96]. Furthermore, numerous animal studies have implicated gene polymorphisms of SERT, as well as genetic and surface transporter expression and function, in the underlying repetitive behaviors and social behavior deficits of ASD, for a review, see [97].
Contributing to the link between serotonin, gut microbiota, and ASD, the BTBR inbred strain has been shown to display (1) reduced SERT density and binding throughout the brain and increased serotonin activity in the hippocampus (a brain region involved in learning, social, and emotional processing, and found to be abnormal in ASD) [98][99][100]; (2) changes in intestinal microbiota associated with slowed GI motility and impaired intestinal serotonin production [48]; and (3) increased sociability following brief exposure to serotonin reuptake inhibitors [98][101] and tryptophan supplementation [102]. Taken together, these studies support the hypothesis that altered gut microbiota are involved in the tryptophan-serotonin metabolic pathway in ASD and provide a framework for future studies aiming to alleviate the GI and, consequently, behavioral symptoms of ASD patients.
A recent study by Fung et al. [103] showed that the gut bacterium Turicibacter sanguinis expresses a neurotransmitter sodium symporter-related protein with sequence and structural homology to mammalian SERT. This microbe imports serotonin through a mechanism that, like its host homologue, is inhibited by the selective serotonin reuptake inhibitor, fluoxetine. Serotonin reduces expression of sporulation factors and membrane transporters in T. sanguinis, which is reversed by fluoxetine exposure. Treating T. sanguinis with serotonin or fluoxetine modulates its competitive colonization in the GI tract of antibiotic-treated mice. In addition, fluoxetine reduces membership of T. sanguinis in the gut microbiota of conventionally colonized mice. One may speculate that genetic variants exist for the microbial SERT-like mechanism and that alterations in the bidirectional host-microbe interactions in tryptophan metabolites play a role in ASD pathophysiology, including gut symptoms.
4.2. Serotonin in the Brain and Relationships with Behavior
Several functional neuroimaging studies indicate that the microbiota-modulated serotonergic system affects neural functioning in ASD. For example, positron emission tomography (PET) ligand studies have observed atypical serotonin functioning throughout the brain that was associated with greater social deficits in individuals with ASD [104][105]. One study found that ASD participants had significantly less binding of thalamic serotonin receptors than age-matched controls and that binding potential in the ASD group was negatively associated with social communication impairments [104]. Another PET study found a global reduction in SERT binding in adults with ASD [103]. Moreover, reduced SERT binding in the anterior and posterior cingulate cortices (ACC; PCC) was associated with impaired social cognition and reduced binding in the thalamus was associated with repetitive behaviors [105]. A more recent PET study found lower serotonin transporter availability in the total gray matter and brainstem of adults with ASD, relative to matched controls [106]. Serotonin transporter availability in the nucleus accumbens, ACC, and putamen was positively correlated with social cognition, indicating that the serotonin transporter may be a marker that can be targeted in pharmacological interventions [106].
Additionally, a polymorphism of the serotonin transporter gene (serotonin-transporter-linked-promoter region; 5-HTTLPR) has been shown to affect brain function in ASD. Short variants of 5-HTTLPR, which reduce serotonin transporter expression [107], are associated with impairments in social communication and interactions in individuals with ASD [108][109]. This is in line with other findings that have linked 5-HTTLPR to the default mode network (DMN), a neural network engaged during passive self-referential cognitive processing and associated with social cognition [110]. Wiggins et al. [111] found that while youth with ASD who had low expressing 5-HTTLPR alleles had stronger posterior-anterior DMN connectivity than those with high expressing genotypes, the opposite was true for TD controls. Thus, resting-state connectivity was shown to be influenced by 5-HTTLPR in a different pattern in ASD than typical controls, suggesting that individuals with low expressing 5-HTTLPR genotypes may comprise a subtype of ASD.
The same researchers showed that ASD youth with low expressing 5-HTTLPR genotypes also had atypical amygdala functioning while performing social tasks [112][113]. During an observational face-processing task, youth with low expressing 5-HTTLPR genotypes failed to show amygdala habituation to repeated observation of sad faces compared to TD controls and ASD youth with high expressing genotypes. Building on these findings, Velasquez et al. [112] showed that when viewing happy faces, the ASD group with the low expressing genotypes had abnormally high rates of functional connectivity between the amygdala and subgenual ACC than ASD individuals with higher expressing genotypes and TD groups of higher and lower expressing genotypes. ASD participants with greater amygdala-subgenual ACC connectivity, key regions involved in emotion arousal and regulation, also showed higher expressing genotypes of 5-HTTLPR and less social dysfunction. Together, these findings indicate that, in ASD, it is possible that some socioemotional functioning during rest and social processing may be influenced by atypicalities in the serotonergic system [112][113]. These results further support the importance of the 5-HTTLPR genotype in examining the heterogeneity of social function in ASD. Nevertheless, a recent meta-analysis found no support for a direct effect of 5-HTTLPR polymorphism on risk of ASD, indicating that patterns across studies may be difficult to find given the heterogeneity of the disorder. Further analyses in larger sample sizes with more homogeneous subgroups of ASD participants may be necessary [114].
Research also suggests that there are group differences between ASD and TD neural responses to serotonin availability. One fMRI study found that increased serotonin (via a selective serotonin reuptake inhibitor) was associated with sustained neural activation in emotion-related brain regions during a negative facial emotion-processing task in ASD adults compared to neurotypical controls, which exhibited an expected habituation response [115]. The authors concluded that homoeostatic control of these regions is altered by serotonin in ASD. Two other fMRI studies found that acute tryptophan depletion (which lowers serotonin levels) was associated with deficits in facial affect [116] and inhibitory processing [117] in ASD. These findings may help explain some of the social and restricted/repetitive behaviors that are hallmarks of the disorder [116][117]. Further, many of the brain regions found to be modulated by the tryptophan depletion have previously been reported to have serotonergic abnormalities in people with ASD (e.g., synthesis differences, lower numbers of receptors and transporters) [116].
Despite the extensive evidence showing alterations in the tryptophan pathway and the serotonergic system in ASD, there is no consensus in current literature on tryptophan availability and its influence on behavior in patients with ASD. While studies have suggested that reduced levels of tryptophan [118][119][120] and decreased tryptophan metabolism are prominent features of ASD [121], a recent study showed both increased and decreased levels of tryptophan in ASD youth [122], which, again, may be related to ASD heterogeneity and marks the need for ASD sub-grouping. The potential roles of oxytocin and other upstream markers in ASD have also been discussed [123] and it is likely that interactions between oxytocin and serotonin may further influence the pathophysiological processes of ASD [124]. More research is necessary to fully understand the complex neurochemical processes underlying the relationship between the BGM system, tryptophan metabolism, and ASD. Below, we discuss specific findings in BGM connections at the structural and functional levels and how changes in the microbiome impact neural networks.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing, Inc.: Arlington, VA, USA, 2013.
- Cowan, C.S.M.; Dinan, T.G.; Cryan, J.F. Annual Research Review: Critical windows—The microbiota-gut-brain axis in neurocognitive development. J. Child Psychol. Psychiatry 2019, 61, 353–371.
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052.
- Logsdon, A.F.; Erickson, M.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood–brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2017, 243, 159–165.
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148.
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938.
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49.
- Mayer, E.A.; Naliboff, B.D.; Craig, A.B. Neuroimaging of the Brain-Gut Axis: From Basic Understanding to Treatment of Functional GI Disorders. Gastroenterology 2006, 131, 1925–1942.
- Mayer, E. The Mind-Gut Connection: How the Hidden Conversation Within Our Bodies Impacts Our Mood, Our Choices, and Our Overall Health. Nature 2016, 536, 146–147.
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522.
- Borre, Y.E.; O’Keeffe, G.; Clarke, G.; Stanton, C.; Dinan, T.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518.
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2019, 11, 135–157.
- Paysour, M.J.; Bolte, A.C.; Lukens, J.R. Crosstalk Between the Microbiome and Gestational Immunity in Autism-Related Disorders. DNA Cell Biol. 2019, 38, 405–409.
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544.
- Flannery, J.; Callaghan, B.; Sharpton, T.; Fisher, P.; Pfeifer, J. Is adolescence the missing developmental link in Microbiome-Gut-Brain axis communication? Dev. Psychobiol. 2019, 61, 783–795.
- Zhuang, L.; Chen, H.; Zhang, S.; Zhuang, J.; Li, Q.; Feng, Z. Intestinal Microbiota in Early Life and Its Implications on Childhood Health. Genom. Proteom. Bioinform. 2019, 17, 13–25.
- Dong, T.S.; Gupta, A. Influence of Early Life, Diet, and the Environment on the Microbiome. Clin. Gastroenterol. Hepatol. 2018, 17, 231–242.
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2016, 6, 1543.
- Reid, B.M.; Horne, R.; Donzella, B.; Szamosi, J.C.; Coe, C.L.; Foster, J.A.; Gunnar, M.R. Microbiota-immune alterations in adolescents following early life adversity: A proof of concept study. Dev. Psychobiol. 2020, 63, 851–863.
- Carlson, A.; Xia, K.; Azcarate-Peril, M.A.; Goldman, B.D.; Ahn, M.; Styner, M.A.; Thompson, A.L.; Geng, X.; Gilmore, J.H.; Knickmeyer, R.C. Infant Gut Microbiome Associated with Cognitive Development. Biol. Psychiatry 2017, 83, 148–159.
- Slykerman, R.F.; Thompson, J.; Waldie, K.; Murphy, R.; Wall, C.; Mitchell, E.A. Antibiotics in the first year of life and subsequent neurocognitive outcomes. Acta Paediatr. 2016, 106, 87–94.
- Slykerman, R.F.; Coomarasamy, C.; Wickens, K.; Thompson, J.M.D.; Stanley, T.V.; Barthow, C.; Kang, J.; Crane, J.; Mitchell, E.A. Exposure to antibiotics in the first 24 months of life and neurocognitive outcomes at 11 years of age. Psychopharmacology 2019, 236, 1573–1582.
- Aatsinki, A.-K.; Lahti, L.; Uusitupa, H.-M.; Munukka, E.; Keskitalo, A.; Nolvi, S.; O’Mahony, S.; Pietilä, S.; Elo, L.L.; Eerola, E.; et al. Gut microbiota composition is associated with temperament traits in infants. Brain Behav. Immun. 2019, 80, 849–858.
- Aatsinki, A.-K.; Kataja, E.-L.; Munukka, E.; Lahti, L.; Keskitalo, A.; Korja, R.; Nolvi, S.; Häikiö, T.; Tarro, S.; Karlsson, H.; et al. Infant fecal microbiota composition and attention to emotional faces. Emotion 2020.
- Christian, L.M.; Galley, J.D.; Hade, E.; Schoppe-Sullivan, S.; Dush, C.K.; Bailey, M. Gut microbiome composition is associated with temperament during early childhood. Brain Behav. Immun. 2014, 45, 118–127.
- Kelsey, C.M.; Prescott, S.; McCulloch, J.A.; Trinchieri, G.; Valladares, T.L.; Dreisbach, C.; Alhusen, J.; Grossmann, T. Gut microbiota composition is associated with newborn functional brain connectivity and behavioral temperament. Brain Behav. Immun. 2020, 91, 472–486.
- Jang, S.-H.; Woo, Y.S.; Lee, S.-Y.; Bahk, W.-M. The Brain–Gut–Microbiome Axis in Psychiatry. Int. J. Mol. Sci. 2020, 21, 7122.
- Lai, M.-C.; Kassee, C.; Besney, R.; Bonato, S.; Hull, L.; Mandy, W.; Szatmari, P.; Ameis, S.H. Prevalence of co-occurring mental health diagnoses in the autism population: A systematic review and meta-analysis. Lancet Psychiatry 2019, 6, 819–829.
- Iglesias-Vázquez, L.; Riba, G.V.G.; Arija, V.; Canals, J. Composition of Gut Microbiota in Children with Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 792.
- Kim, Y.-K.; Shin, C. The Microbiota-Gut-Brain Axis in Neuropsychiatric Disorders: Pathophysiological Mechanisms and Novel Treatments. Curr. Neuropharmacol. 2018, 16, 559–573.
- Yang, J.; Fu, X.; Liao, X.; Li, Y. Effects of gut microbial-based treatments on gut microbiota, behavioral symptoms, and gastrointestinal symptoms in children with autism spectrum disorder: A systematic review. Psychiatry Res. 2020, 293, 113471.
- Al-Beltagi, M. Autism medical comorbidities. World J. Clin. Pediatr. 2021, 10, 15–28.
- Dan, Z.; Mao, X.; Liu, Q.; Guo, M.; Zhuang, Y.; Liu, Z.; Chen, K.; Chen, J.; Xu, R.; Tang, J.; et al. Altered gut microbial profile is associated with abnormal metabolism activity of Autism Spectrum Disorder. Gut Microbes 2020, 11, 1246–1267.
- Hughes, H.; Rose, D.; Ashwood, P. The Gut Microbiota and Dysbiosis in Autism Spectrum Disorders. Curr. Neurol. Neurosci. Rep. 2018, 18, 81.
- Kang, D.-W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 2018, 49, 121–131.
- Needham, B.D.; Adame, M.D.; Serena, G.; Rose, D.R.; Preston, G.M.; Conrad, M.C.; Campbell, A.S.; Donabedian, D.H.; Fasano, A.; Ashwood, P.; et al. Plasma and Fecal Metabolite Profiles in Autism Spectrum Disorder. Biol. Psychiatry 2020, 89, 451–462.
- Parracho, H.M.R.T.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991.
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal microbiota in children with autism in Slovakia. Physiol. Behav. 2015, 138, 179–187.
- Mortera, S.L.; Vernocchi, P.; Basadonne, I.; Zandonà, A.; Chierici, M.; Durighello, M.; Marzano, V.; Gardini, S.; Gasbarrini, A.; Urbani, A.; et al. A metaproteomic-based gut microbiota profiling in children affected by autism spectrum disorders. J. Proteom. 2021, 251, 104407.
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521.
- Sherwin, E.; Bordenstein, S.R.; Quinn, J.L.; Dinan, T.G.; Cryan, J.F. Microbiota and the social brain. Science 2019, 366.
- Archie, E.A.; Tung, J. Social behavior and the microbiome. Curr. Opin. Behav. Sci. 2015, 6, 28–34.
- Münger, E.; Montiel-Castro, A.J.; Langhans, W.; Pacheco-López, G. Reciprocal Interactions between Gut Microbiota and Host Social Behavior. Front. Integr. Neurosci. 2018, 12, 21.
- Needham, B.; Tang, W.; Wu, W.-L. Searching for the gut microbial contributing factors to social behavior in rodent models of autism spectrum disorder. Dev. Neurobiol. 2018, 78, 474–499.
- Buffington, S.A.; di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775.
- Kamimura, I.; Kaneko, R.; Morita, H.; Mogi, K.; Kikusui, T. Microbial colonization history modulates anxiety-like and complex social behavior in mice. Neurosci. Res. 2021, 168, 64–75.
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148.
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Pérez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178.
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463.
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Low Relative Abundances of the Mucolytic Bacterium Akkermansia muciniphila and Bifidobacterium spp. in Feces of Children with Autism. Appl. Environ. Microbiol. 2011, 77, 6718–6721.
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274.
- Vuong, H.E.; Hsiao, E.Y. Emerging Roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiatry 2016, 81, 411–423.
- Xu, M.; Xu, X.; Li, J.; Li, F. Association between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473.
- Sharon, G.; Cruz, N.J.; Kang, D.-W.; Gandal, M.J.; Wang, B.; Kim, Y.-M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17.
- Xiao, L.; Yan, J.; Yang, T.; Zhu, J.; Li, T.; Wei, H.; Chen, J. Fecal Microbiome Transplantation from Children with Autism Spectrum Disorder Modulates Tryptophan and Serotonergic Synapse Metabolism and Induces Altered Behaviors in Germ-Free Mice. mSystems 2021, 6.
- FAO/WHO. Evaluation of Health and Nutritional Properties of Powder Milk and Live Lactic Acid Bacteria. Food and Agriculture Organization of the United Nations and World Health Organization Expert Consultation Report. 2001. Available online: https://www.fao.org/tempref/docrep/fao/meeting/009/y6398e.pdf (accessed on 5 November 2021).
- Isolauri, E.; Salminen, S.; Rautava, S. Early Microbe Contact and Obesity Risk. J. Pediatr. Gastroenterol. Nutr. 2016, 63, S3–S5.
- Pärtty, A.; Rautava, S.; Kalliomäki, M. Probiotics on Pediatric Functional Gastrointestinal Disorders. Nutrients 2018, 10, 1836.
- Patusco, R.; Ziegler, J. Role of Probiotics in Managing Gastrointestinal Dysfunction in Children with Autism Spectrum Disorder: An Update for Practitioners. Adv. Nutr. 2018, 9, 637–650.
- Wilkins, T.; Sequoia, J. Probiotics for Gastrointestinal Conditions: A Summary of the Evidence. Am. Fam. Physician 2017, 96, 170–178.
- Haddad, F.L.; Patel, S.V.; Schmid, S. Maternal Immune Activation by Poly I:C as a preclinical Model for Neurodevelopmental Disorders: A focus on Autism and Schizophrenia. Neurosci. Biobehav. Rev. 2020, 113, 546–567.
- Liu, Y.-W.; Liong, M.T.; Chung, Y.-C.E.; Huang, H.-Y.; Peng, W.-S.; Cheng, Y.-F.; Lin, Y.-S.; Wu, Y.-Y.; Tsai, Y.-C. Effects of Lactobacillus plantarum PS128 on Children with Autism Spectrum Disorder in Taiwan: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2019, 11, 820.
- Meyyappan, A.C.; Forth, E.; Wallace, C.J.K.; Milev, R. Effect of fecal microbiota transplant on symptoms of psychiatric disorders: A systematic review. BMC Psychiatry 2020, 20, 299.
- Kang, D.-W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10.
- Kang, D.-W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821.
- Adams, J.B.; Vargason, T.; Kang, D.-W.; Krajmalnik-Brown, R.; Hahn, J. Multivariate Analysis of Plasma Metabolites in Children with Autism Spectrum Disorder and Gastrointestinal Symptoms Before and After Microbiota Transfer Therapy. Processes 2019, 7, 806.
- Qureshi, F.; Adams, J.; Hanagan, K.; Kang, D.-W.; Krajmalnik-Brown, R.; Hahn, J. Multivariate Analysis of Fecal Metabolites from Children with Autism Spectrum Disorder and Gastrointestinal Symptoms before and after Microbiota Transfer Therapy. J. Pers. Med. 2020, 10, 152.
- Chen, K.; Fu, Y.; Wang, Y.; Liao, L.; Xu, H.; Zhang, A.; Zhang, J.; Fan, L.; Ren, J.; Fang, B. Therapeutic Effects of the In Vitro Cultured Human Gut Microbiota as Transplants on Altering Gut Microbiota and Improving Symptoms Associated with Autism Spectrum Disorder. Microb. Ecol. 2020, 80, 475–486.
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794.
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113.
- Gao, K.; Mu, C.-L.; Farzi, A.; Zhu, W.-Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2019, 11, 709–723.
- de Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; de Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal Microbiota and Metabolome of Children with Autism and Pervasive Developmental Disorder Not Otherwise Specified. PLoS ONE 2013, 8, e76993.
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47.
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An In Silico Analysis. Front. Neurosci. 2019, 13, 1365.
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973.
- Diémé, B.; Mavel, S.; Blasco, H.; Tripi, G.; Bonnet-Brilhault, F.; Malvy, J.; Bocca, C.; Andres, C.R.; Nadal-Desbarats, L.; Emond, P. Metabolomics Study of Urine in Autism Spectrum Disorders Using a Multiplatform Analytical Methodology. J. Proteome Res. 2015, 14, 5273–5282.
- Olesova, D.; Galba, J.; Piestansky, J.; Celusakova, H.; Repiska, G.; Babinska, K.; Ostatnikova, D.; Katina, S.; Kovac, A. A Novel UHPLC-MS Method Targeting Urinary Metabolomic Markers for Autism Spectrum Disorder. Metabolites 2020, 10, 443.
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412.
- Savino, R.; Carotenuto, M.; Polito, A.; di Noia, S.; Albenzio, M.; Scarinci, A.; Ambrosi, A.; Sessa, F.; Tartaglia, N.; Messina, G. Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway. Brain Sci. 2020, 10, 631.
- Lim, C.K.; Essa, M.M.; Martins, R.D.P.; Lovejoy, D.B.; Bilgin, A.; Waly, M.; Al-Farsi, Y.M.; Al-Sharbati, M.; Al-Shaffae, M.A.; Guillemin, G. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity. Autism Res. 2015, 9, 621–631.
- Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Saugstad, O.D.; Ormstad, H. Kynurenine Pathway in Autism Spectrum Disorders in Children. Neuropsychobiology 2017, 76, 82–88.
- Luna, R.A.; Oezguen, N.; Balderas, M.; Venkatachalam, A.; Runge, J.K.; Versalovic, J.; Veenstra-VanderWeele, J.; Anderson, G.M.; Savidge, T.; Williams, K.C. Distinct Microbiome-Neuroimmune Signatures Correlate with Functional Abdominal Pain in Children with Autism Spectrum Disorder. Cell. Mol. Gastroenterol. Hepatol. 2016, 3, 218–230.
- Jenkins, T.A.; Nguyen, J.C.D.; Polglaze, K.E.; Bertrand, P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56.
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48.
- Terry, N.; Margolis, K.G. Serotonergic Mechanisms Regulating the GI Tract: Experimental Evidence and Therapeutic Relevance. Gastrointest. Pharmacol. 2016, 239, 319–342.
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276.
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 8952520.
- Gabriele, S.; Sacco, R.; Persico, A.M. Blood serotonin levels in autism spectrum disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2014, 24, 919–929.
- Muller, C.; Anacker, A.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2015, 321, 24–41.
- Anderson, G.M.; Horne, W.C.; Chatterjee, D.; Cohen, D.J. The Hyperserotonemia of Autism. Ann. N. Y. Acad. Sci. 1990, 600, 331–340.
- Lim, J.S.; Lim, M.Y.; Choi, Y.; Ko, G. Modeling environmental risk factors of autism in mice induces IBD-related gut microbial dysbiosis and hyperserotonemia. Mol. Brain 2017, 10, 14.
- Tanaka, M.; Sato, A.; Kasai, S.; Hagino, Y.; Kotajima-Murakami, H.; Kashii, H.; Takamatsu, Y.; Nishito, Y.; Inagaki, M.; Mizuguchi, M.; et al. Brain hyperserotonemia causes autism-relevant social deficits in mice. Mol. Autism 2018, 9, 60.
- Marler, S.; Ferguson, B.J.; Lee, E.B.; Peters, B.; Williams, K.C.; McDonnell, E.; Macklin, E.A.; Levitt, P.; Gillespie, C.H.; Anderson, G.M.; et al. Brief Report: Whole Blood Serotonin Levels and Gastrointestinal Symptoms in Autism Spectrum Disorder. J. Autism Dev. Disord. 2015, 46, 1124–1130.
- Israelyan, N.; Margolis, K.G. Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders. Pharmacol. Res. 2018, 132, 1–6.
- Veenstra-VanderWeele, J.; Muller, C.L.; Iwamoto, H.; Sauer, J.E.; Owens, W.A.; Shah, C.R.; Cohen, J.; Mannangatti, P.; Jessen, T.; Thompson, B.; et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. USA 2012, 109, 5469–5474.
- Abdulamir, H.A.; Rasheed, O.F.A.; Abdulghani, E.A. Serotonin and serotonin transporter levels in autistic children. Saudi Med. J. 2018, 39, 487–494.
- Garbarino, V.R.; Gilman, T.L.; Daws, L.C.; Gould, G.G. Extreme enhancement or depletion of serotonin transporter function and serotonin availability in autism spectrum disorder. Pharmacol. Res. 2018, 140, 85–99.
- Gould, G.G.; Hensler, J.G.; Burke, T.F.; Benno, R.H.; Onaivi, E.S.; Daws, L.C. Density and function of central serotonin (5-HT) transporters, 5-HT1A and 5-HT2A receptors, and effects of their targeting on BTBR T+tf/J mouse social behavior. J. Neurochem. 2010, 116, 291–303.
- Gould, G.G.; Burke, T.F.; Osorio, M.D.; Smolik, C.M.; Zhang, W.Q.; Onaivi, E.S.; Gu, T.-T.; DeSilva, M.N.; Hensler, J.G. Enhanced novelty-induced corticosterone spike and upregulated serotonin 5-HT1A and cannabinoid CB1 receptors in adolescent BTBR mice. Psychoneuroendocrinology 2013, 39, 158–169.
- Banker, S.M.; Gu, X.; Schiller, D.; Foss-Feig, J.H. Hippocampal contributions to social and cognitive deficits in autism spectrum disorder. Trends Neurosci. 2021, 44, 793–807.
- Chadman, K.K. Fluoxetine but not risperidone increases sociability in the BTBR mouse model of autism. Pharmacol. Biochem. Behav. 2011, 97, 586–594.
- Zhang, W.Q.; Smolik, C.M.; Barba-Escobedo, P.A.; Gamez, M.; Sanchez, J.J.; Javors, M.A.; Daws, L.C.; Gould, G.G. Acute dietary tryptophan manipulation differentially alters social behavior, brain serotonin and plasma corticosterone in three inbred mouse strains. Neuropharmacology 2014, 90, 1–8.
- Fung, T.C.; Vuong, H.E.; Luna, C.D.G.; Pronovost, G.N.; Aleksandrova, A.; Riley, N.G.; Vavilina, A.; McGinn, J.; Rendon, T.; Forrest, L.R.; et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat. Microbiol. 2019, 4, 2064–2073.
- Beversdorf, D.Q.; Nordgren, R.E.; Bonab, A.A.; Fischman, A.J.; Weise, S.B.; Dougherty, D.D.; Felopulos, G.J.; Zhou, F.C.; Bauman, M.L. 5-HT2Receptor Distribution Shown by Setoperone PET in High-Functioning Autistic Adults. J. Neuropsychiatry Clin. Neurosci. 2012, 24, 191–197.
- Nakamura, K.; Sekine, Y.; Ouchi, Y.; Tsujii, M.; Yoshikawa, E.; Futatsubashi, M.; Tsuchiya, K.; Sugihara, G.; Iwata, Y.; Suzuki, K.; et al. Brain Serotonin and Dopamine Transporter Bindings in Adults with High-Functioning Autism. Arch. Gen. Psychiatry 2010, 67, 59–68.
- Andersson, M.; Tangen, Ä.; Farde, L.; Bölte, S.; Halldin, C.; Borg, J.; Lundberg, J. Serotonin transporter availability in adults with autism—a positron emission tomography study. Mol. Psychiatry 2020, 26, 1647–1658.
- Lesch, K.-P.; Bengel, D.; Heils, A.; Sabol, S.Z.; Greenberg, B.D.; Petri, S.; Benjamin, J.; Müller, C.R.; Hamer, D.H.; Murphy, D.L. Association of Anxiety-Related Traits with a Polymorphism in the Serotonin Transporter Gene Regulatory Region. Science 1996, 274, 1527–1531.
- Brune, C.W.; Kim, S.-J.; Salt, J.; Leventhal, B.L.; Lord, C.; Cook, E. 5-HTTLPR Genotype-Specific Phenotype in Children and Adolescents with Autism. Am. J. Psychiatry 2006, 163, 2148–2156.
- Tordjman, S.; Gutknecht, L.; Carlier, M.; Spitz, E.; Antoine, C.; Slama, F.; Carsalade, V.; Cohen, D.J.; Ferrari, P.; Roubertoux, P.L.; et al. Role of the serotonin transporter gene in the behavioral expression of autism. Mol. Psychiatry 2001, 6, 434–439.
- Buckner, R.L.; Andrews-Hanna, E.J.R.; Schactera, D.L. The Brain’s Default Network: Anatomy, Function, and Relevance to Disease. Ann. N. Y. Acad. Sci. 2008, 1124, 1–38.
- Wiggins, J.L.; Peltier, S.J.; Bedoyan, J.K.; Carrasco, M.; Welsh, R.C.; Martin, D.M.; Lord, C.; Monk, C.S. The impact of serotonin transporter genotype on default network connectivity in children and adolescents with autism spectrum disorders. NeuroImage Clin. 2012, 2, 17–24.
- Velasquez, F.; Wiggins, J.L.; Mattson, W.I.; Martin, D.M.; Lord, C.; Monk, C.S. The influence of 5-HTTLPR transporter genotype on amygdala-subgenual anterior cingulate cortex connectivity in autism spectrum disorder. Dev. Cogn. Neurosci. 2016, 24, 12–20.
- Wiggins, J.L.; Swartz, J.; Martin, D.M.; Lord, C.; Monk, C.S. Serotonin transporter genotype impacts amygdala habituation in youth with autism spectrum disorders. Soc. Cogn. Affect. Neurosci. 2013, 9, 832–838.
- Wang, H.; Yin, F.; Gao, J.; Fan, X. Association Between 5-HTTLPR Polymorphism and the Risk of Autism: A Meta-Analysis Based on Case-Control Studies. Front. Psychiatry 2019, 10.
- Wong, N.M.L.; Findon, J.L.; Wichers, R.H.; Giampietro, V.; Stoencheva, V.; Murphy, C.M.; Blainey, S.; Ecker, C.; Murphy, D.G.; McAlonan, G.M.; et al. Serotonin differentially modulates the temporal dynamics of the limbic response to facial emotions in male adults with and without autism spectrum disorder (ASD): A randomised placebo-controlled single-dose crossover trial. Neuropsychopharmacology 2020, 45, 2248–2256.
- Daly, E.M.; Deeley, Q.; Ecker, C.; Craig, M.; Hallahan, B.; Murphy, C.M.; Johnston, P.; Spain, D.; Gillan, N.; Brammer, M.; et al. Serotonin and the Neural Processing of Facial Emotions in Adults with Autism. Arch. Gen. Psychiatry 2012, 69, 1003.
- Daly, E.M.; Ecker, C.; Hallahan, B.; Deeley, Q.; Craig, M.; Murphy, C.M.; Johnston, P.; Spain, D.; Gillan, N.; Gudbrandsen, M.; et al. Response inhibition and serotonin in autism: A functional MRI study using acute tryptophan depletion. Brain 2014, 137, 2600–2610.
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. 2011, 8, 34.
- Kałuzna-Czaplinska, J.; Michalska, M.; Rynkowski, J. Determination of tryptophan in urine of autistic and healthy children by gas chromatography/mass spectrometry. Med. Sci. Monit. 2010, 16, CR488–CR492.
- Naushad, S.M.; Jain, J.M.N.; Prasad, C.K.; Naik, U.; Akella, R.R.D. Autistic children exhibit distinct plasma amino acid profile. Indian J. Biochem. Biophys. 2013, 50, 474–478.
- Boccuto, L.; Chen, C.-F.; Pittman, A.R.; Skinner, C.D.; McCartney, H.J.; Jones, K.; Bochner, B.R.; Stevenson, R.E.; Schwartz, C.E. Decreased tryptophan metabolism in patients with autism spectrum disorders. Mol. Autism 2013, 4, 16.
- Kałużna-Czaplińska, J.; Jóźwik-Pruska, J.; Chirumbolo, S.; Bjørklund, G. Tryptophan status in autism spectrum disorder and the influence of supplementation on its level. Metab. Brain Dis. 2017, 32, 1585–1593.
- Ooi, Y.P.; Weng, S.-J.; Kossowsky, J.; Gerger, H.; Sung, M. Oxytocin and Autism Spectrum Disorders: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Pharmacopsychiatry 2016, 50, 5–13.
- Maes, M.; Anderson, G.; Medina, S.R.B.; Seo, M.; Ojala, J.O. Integrating Autism Spectrum Disorder Pathophysiology: Mitochondria, Vitamin A, CD38, Oxytocin, Serotonin and Melatonergic Alterations in the Placenta and Gut. Curr. Pharm. Des. 2020, 25, 4405–4420.
More
Information
Subjects:
Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
17 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No