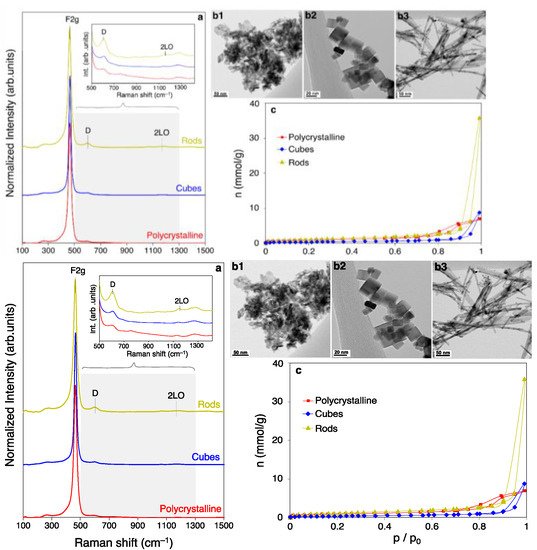
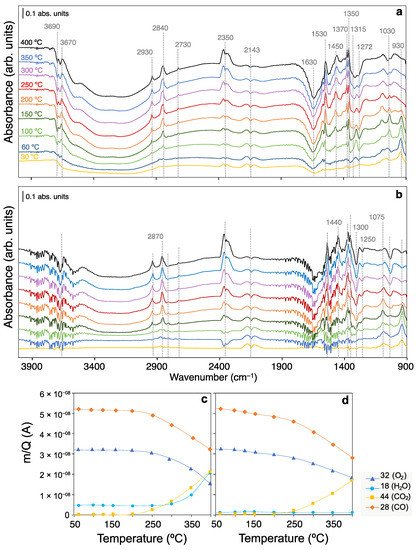
4.1. Surface Coverage Studies in CO-PROX Conditions
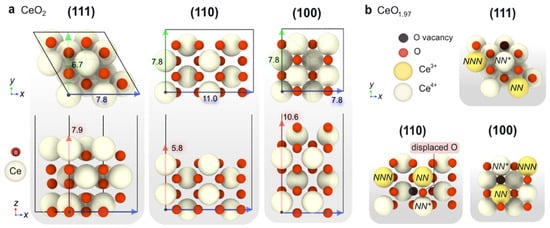
Firstly, the resting state of each of the ceria surfaces under relevant CO-PROX conditions was determined. Upon the dissociative adsorption of H
2, two OH groups are formed
[69] leading to the one-electron reduction of two Ce
4+ surface sites to Ce
3+. Since theoretical calculations have demonstrated that H
2 dissociation on ceria is thermodynamically driven at relatively low temperatures
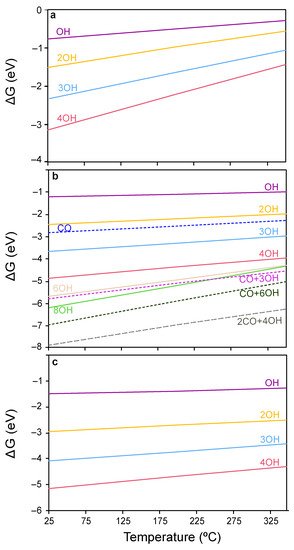
[70][71], the H coverage was assessed herein by calculating the energies of ceria surfaces covered with different concentrations of OH groups. The coverages explored and their corresponding relative Gibbs energies in the temperature range of 25–350 °C are represented in
Figure 4a–c. As can be observed, throughout the temperature window of interest in CO-PROX (i.e., 25–200 °C), ceria surfaces are predicted to be covered by H in the form of OH groups. In particular, the four surface O atoms in CeO
2(111) and CeO
2(100) are fully hydrogenated (4 OH), while the eight surface oxygens in CeO
2(110) are terminated with 4 OHs and 2 CO moieties strongly stabilized as carbonates (see below).
Figure 4. Coverage analysis on the ceria surfaces (a) (111), (b) (110) and (c) (100). Solid lines denote OH coverages, while dotted lines represent mixed coverages of OH and CO groups.
With regards to CO, the Gibbs adsorption energies of this species on the clean (111) and (100) surfaces are relatively weak (i.e., −0.18 and −0.33 eV, respectively) compared to hydrogen. On the contrary, CO binds very strongly on the (110) surface as a carbonate-like species with an energy of –3.66 eV, in good agreement with the literature
[72][73][74][75], leading to the reduction of two neighboring Ce
4+ cations.
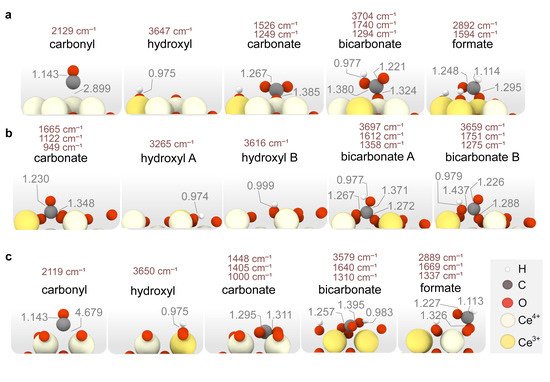
Figure 5 shows the most stable configurations of the adsorbates found in CO oxidation conditions on each stoichiometric ceria surface. Since the coverage analysis reveals that OHs populate the ceria surfaces in the presence of H
2, the Gibbs adsorption energies of *CO
x adsorbates at different temperatures and OH coverages were computed. Interestingly, the binding modes of *CO
x species remain unaltered in the presence of OH coverage despite the local Ce reduction induced by H
2 dissociation leads to stronger *CO
x–ceria interactions. Ceria surfaces may present two types of Lewis basic sites for CO
x adsorption, namely surface oxygens and hydroxyls
[76]. Whilst OHs are deemed as weaker base centers
[77], their presence on the surface increases the Lewis basicity of surface oxygens due to the coexistence of reduced Ce
3+ cations. Thus, CO
2 adsorption on the hydroxylated surfaces gives rise to bicarbonates (O*COOH), while CO
2 adsorption on O sites leads to carbonates (O*COO). On the other hand, the adsorption of CO on OH-covered ceria surfaces can lead to formates (O*CHO), which are very stable.
Figure 5. Representation of the most energetically favorable adsorption modes for CO, CO2 and H2 on stoichiometric (a) CeO2(111), (b) CeO2(110), (c) CeO2(100). Calculated vibrational frequencies (in cm−1) and relevant distances (in Å) are also shown.
DFT calculations also reveal that the type of surface carbonate species varies with the surface termination. In particular, carbonates formed upon CO2 adsorption on CeO2(111) display a monodentate mode, while on CeO2(100) they adopt a multidentate geometry with a much lower relative energy (i.e., −1.96 vs. −0.46 eV). Similarly, CO2 adsorption on the clean (110) surface results in a monodentate carbonate with a relative energy of −1.31 eV (not shown in Figure 5 since the carbonate formed upon CO adsorption has a remarkably greater stability).
As regards to CO, this species merely physisorbs on top of Ce in CeO
2(111), while it forms a bridged Ce-CO carbonyl on CeO
2(100), and a strongly bound bidentate carbonate on CeO
2(110). In addition, the most stable bicarbonate binding modes vary on the different surfaces, being monodentate in (111), bidentate in (110) and multidentate in (100). Finally, from the calculated CO
2 adsorption energies on the clean surfaces, we can conclude that the strength of the Lewis basic sites increases according to the trend (111) < (110) < (100), in agreement with the literature
[77]. Our calculations also indicate that OH coverages hinder CO
2 adsorption as carbonates on the ceria surfaces. An exception, however, is the CO adsorption on (110) since the formation of a carbonate species provides a larger stabilization than the OH coverage, as observed in
Figure 4b (4 OHs vs. CO + 3 OHs). Altogether, DFT calculations indicate that ceria surfaces will be hydroxylated under CO-PROX conditions and that no surface oxygens may be available. In the next section, the CO oxidation mechanism is studied on both the clean and OH-covered ceria surfaces representative of CO-TOX (H
2-free CO oxidation) and CO-PROX conditions. Since CeO
2(100) is the least exposed surface in representative polycrystalline ceria samples, given it has the highest surface energy, our further reactivity studies placed more focus on the (111) and (110) facets.
4.2. CO Oxidation on CeO2 in CO-PROX Atmosphere
To discern the potential catalytic effect of H2 on CO oxidation, two scenarios are contemplated in the present study, namely a ceria clean surface and a hydroxylated surface.
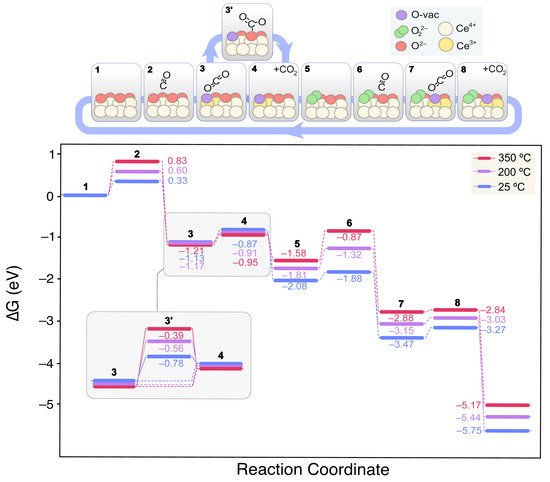
Figure 6 shows the lowest energy mechanism found for CO oxidation on the clean CeO2(111) surface.
Figure 6. Calculated Gibbs energy diagram for the CO oxidation mechanism on a stoichiometric CeO2(111) clean surface at different experimental temperatures. The structures of the different reaction intermediates are depicted at the top of the diagram.
On the clean CeO2(111) surface, CO remains physisorbed at 2.899 Å from a surface Ce4+ ion (2). The CO Gibbs binding energy increases with temperature from 0.33 to 0.83 eV within the range of 25 to 350 °C. Next (3), CO diffuses from the top-Ce position to bind a surface O2− leading to CO2 and an oxygen vacancy (VO). This process is exergonic by −1.46 eV (at 25 °C) given the increased stability of CO2 with regards to CO. The resulting CO2 is physisorbed on the boundaries of the recently formed VO. Subsequently, it can be either directly desorbed to the gas phase (4) or chemisorbed on another surface O2− giving rise to a carbonate CO32− species (3′). Although at room temperature these processes might be in equilibrium, the release of CO2 to the gas phase will prevail at higher temperatures. Next, the O2 adsorption on the VO leads to a peroxide (O22−) intermediate (5), which restitutes the oxidized redox state of ceria, lowering the energy by −1.21 eV at 25 °C. From this state, a new CO molecule could interact with the surface as in (2). The CO binding energy on this surface (6), however, does not change when compared to that on the clean slab (2), and no interaction between *CO(ads) and *O22−(ads) was observed. On the contrary, as in (3), the interaction between the O2–(lat) bound to CO leads to the release of *CO2(ads) and the concomitant formation of a new VO. Finally, the loosely bound O from the peroxide group migrates to the neighboring VO and refills it recovering the original state and closing the catalytic cycle (9).
In this reaction scheme, the endergonic steps are essentially the weak CO adsorptions, whereas the overall process is thermodynamically very favored. In fact, the interaction is so weak that it is also claimed that CO oxidation may proceed via Eley–Rideal mechanism on ceria, involving a CO from the gas phase reacting with surface O
[78]. This is corroborated by our CI-NEB results, where no transition state (TS) could be found for this process, but a V-bent structure with a partially formed C–O–O bond.
Based on the coverage analysis presented in Figure 4a, the CeO2(111) slab is fully hydroxylated under CO-PROX conditions, meaning that all surface oxygens are hydroxylated. However, in contrast to O2− our results showed that CO is not capable to directly bind to OH. Herein, we opted to study a 2 OH-covered surface to model the ceria in the CO-PROX reaction so that O2− sites are available for the reaction. Nevertheless, we modelled the reaction mechanism under different coverages to assess the best pathway and rule out a OH coverage dependence. According to this analysis, the presence of a third OH group systematically increases the stability of the COx intermediates on the surface, while the relative energy difference between steps is not significantly affected. Thus, although high OH coverages impede the CO-surface interaction by extensively blocking the active O2−, the residual freely-exposed O2− sides will display an enhanced basicity.
To deplete the stable OH groups on CeO2(111), sufficient energy must be provided for these OH to recombine giving rise to H2O and a VO. Then, new available O2− sites would be formed by the subsequent refilling by O2 from the gas phase. The energy for OH recombination was calculated to be endergonic by 1.68 eV at 25 °C, and 1.30 eV at 200 °C. Thus, at temperatures above 200 °C it is expected that some of the hydroxyl groups recombine leading to oxygen vacancies and increasing lattice defects.
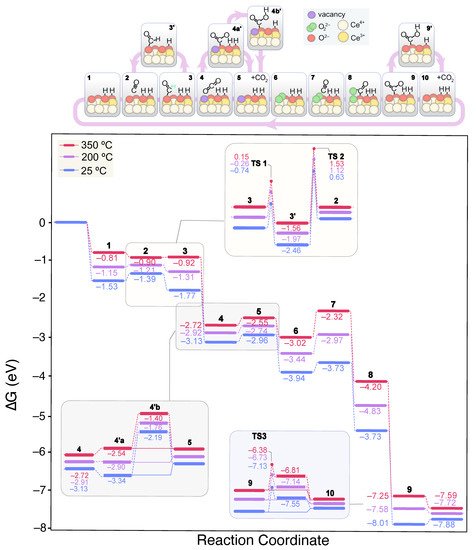
Figure 7 shows the lowest energy mechanism found for CO oxidation on the 2 OH-CeO
2(111) surface. In contrast to the clean, fully oxidized surface, CO can form a carboxylate (*OCO
2−) on the 2 OH-covered surface, a V-shaped CO
2 molecule with a O–C–O angle of 134.3° (structure 3 in
Figure 7). The formation of carboxylates, also known as carbonites, is thermodynamically driven even at 25 °C by the adsorption of CO on a surface oxygen according to: CO(gas) + O
2−(lat) → CO
22−(ads). This process was modeled by means of CI-NEB calculations, displaying two low energy transition states of 0.24 and 0.33 eV, structures corresponding to CO approaching and C–O
2−(lat) bond formation. Carboxylates have been found on pre-reduced ceria surfaces
[79], and naturally lead to carbonates (CO
32−) either by further interaction with surface oxygen or with gaseous O
2. Carboxylates can also serve as precursors of formate species when neighboring OH groups are present
(3′).
Figure 7. Calculated Gibbs energy diagram for the CO oxidation mechanism on a stoichiometric CeO2(111) surface covered with 2 OH groups at different experimental temperatures. The structures of the different reaction intermediates are depicted at the top of the diagram.
TS1 in
Figure 7 refers to the formation of formate from carboxylate, which according to our calculations, requires an activation energy of ca. 1 eV. This low activation energy for formate formation, is consistent with these species being observed by DRIFTS at temperatures as low as 60 °C. In fact, formates are very stable species with relative Gibbs energies of −1.56, −2.22 and −3.02 eV at 350 °C when surrounded by one, two or three OH groups, respectively. Even at high OH coverages, formates are significantly stabilized as their evolution towards CO(ads) + *OH is very energy demanding, as recently showed in our mechanistic study of the CO
2 methanation reaction on ceria
[80]. Hence, we predict formates to accumulate on ceria for a broad temperature window, as observed by DRIFTS. Even without H
2 in the reactant stream (CO-TOX conditions), formates are detected experimentally during CO oxidation around the same temperatures than in CO-PROX conditions.
Calculations also predict that carboxylates do not form on the clean CeO
2(111) surface, but they can be generated in the presence of a V
O and OH groups, which are typically found in real ceria surfaces. Since formates evolution is hindered and independent of the OH coverage during CO oxidation, we conclude that these species are mere spectators. Alternatively, carboxylates can evolve as CO
2, leaving behind a surface V
O (
4). The generated CO
2 can then be readsorbed on a O
2− site to give rise to a carbonate
(4a’), a process that is exergonic by ca. 0.2 eV at room temperature. In contrast to the clean surface, carbonate formation competes with CO
2 desorption
(5). However, since OH groups tend to stabilize carbonates on the surface , the presence of a new OH group is expected to favor the presence of carbonates over CO
2 desorption. This situation is reversed at 200 °C and higher temperatures, and CO
2 desorption prevails regardless of the coverage. While on the surface, carbonates can also be hydrogenated to bicarbonates via H transfer from the coverage
(4b’), which are less stable. As previously discussed by some of us and others
[30][31], a higher bicarbonates-to-carbonates ratio enhances the CO-PROX activity in CuO/CeO
2 catalysts. Furthermore, the depletion of the hydroxyl band correlates with the bicarbonate formation rate. However, according to our DFT calculations, this process has associated a TS with an energy barrier of 0.87 eV (TS3) at room temperature, and therefore, is likely to be able to compete with CO
2 desorption. When comparing the calculated barriers at higher OH coverages, we observe that bicarbonate formation is slightly facilitated. Altogether, the bicarbonate route does not seem to be feasible on the (111) surface based on the calculated reaction thermodynamics and kinetics. In fact, this would not be a limiting step in metal-free ceria catalysts since much higher temperatures are needed to overcome the CO oxidation onset. However, as depicted by the energies of CO
x + OH adsorbates, the (100) surface is the most prone to accumulate carbonates as these species are remarkably stabilized. Hence, in this minor facet, we envision that bicarbonates may have a key role in the CO-PROX catalysis at low temperatures.
Regardless of the reaction pathway, COx is released as CO2 to the gas phase, followed by the regeneration of the VO by a gas O2 molecule (6). To shed light into this process, we investigated all the possible reaction pathways. Firstly, we considered an additional CO adsorbed on a surface still containing the carbonate and the VO, while leaving the O2 uptake to the end of the CO oxidation cycles. This reaction pathway was found to be higher in energy all along the reaction cycle since O2 stabilizes the reaction intermediates. Secondly, we explored the VO restoration by O2 whilst maintaining on surface the carbonate, which is a dead-end unless the carbonate releases the active site since CO(g) and O22− do not interact in these conditions. Thus, the mechanism proposed in Figure 7 is the lowest energy pathway encountered. The second CO molecule finds a free O2− site after CO2 desorption. Instead of forming a carboxylate, we found that a O–O–COO intermediate is stabilized as a result of the CO–O22– interaction (8). This intermediate has an elongated O–O bond at 1.497 Å, indicative of the O22− dissociation degree, while the C–O bond distance is 1.436 Å and the lattice O is displaced upwards from its ground position. Subsequently, this structure leads to a bidentate carbonate and a surface oxygen (9) via the rearrangement of the surface and the O–O bond cleavage. The desorption of the resulting carbonate regenerates the initial state of the catalyst.
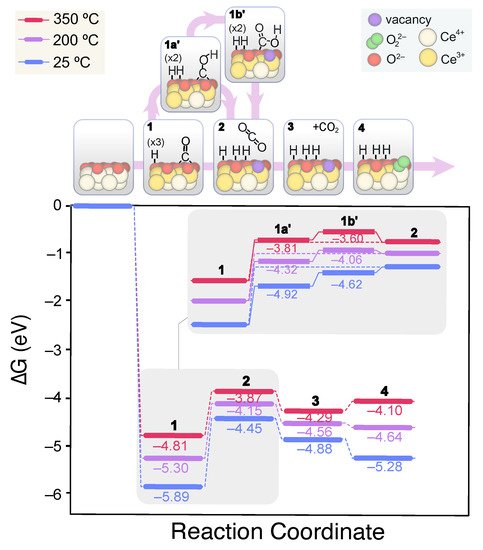
Unlike the (111) surface, CO adsorption on the (110) facet leads to highly stable bidentate carbonates in presence or absence of OH groups. Importantly, CO adsorption involves the reduction of two surface Ce4+ cations and the oxidation of C. Nevertheless, for catalytic CO oxidation, the product must be desorbed and the active site regenerated. The carbonates desorption to CO2 involves an up-hill process of 2.32 eV, whereas their protonation to less stable bicarbonates goes through a high energy TS of 3.7 eV. On the (110) surface, two types of bicarbonates can be formed, labeled herein as type A and B. The former are bound in a bidentate fashion and are more stable than the B-type by ca. –0.3 eV (see Figure 5b). The formation of type B bicarbonates from carbonates is more hampered due to the lower accessibility of the O being protonated. However, the evolution of type A bicarbonates to CO2 + OH involves more rearrangement and thus higher energies due to the greater distance of H+ to the surface. Hence, we propose that type A bicarbonates evolve to the B type through a low energy (i.e., 1.25 eV) H migration, from which CO2 can be released more easily. These insights are depicted in Figure 8, which displays the obtained lower energy CO oxidation mechanism computed for the 3 OH-covered surface. As in the case of CeO2(111), with the OH coverage, the lesser the barrier energies for the overall CO oxidation process, though this cannot be attributed to the more facile bicarbonates formation.
Figure 8. Calculated Gibbs energy diagram for the CO oxidation mechanism on a stoichiometric CeO2(110) surface covered with 3 OH groups at different experimental temperatures. The structures of the different reaction intermediates are depicted at the top of the diagram.
Despite being the least stable surface, the (100) facet might also contribute to the CO oxidation process in polycrystalline ceria catalysts. According to our calculations, CO adsorption on (100) is weak, as in the case of (111), and CO oxidation proceeds via a similar reaction mechanism. However, the (100) surface is less dense, and its more open structure is able to stabilize multidentate carbonates whose desorption is strongly hampered. Thus, the (100) surface may reabsorb and retain the released CO2 either through this or other catalytically active surfaces, increasing the accumulation of product-type adsorbates and leading to surface poisoning. In this case, the protonation of multidentate carbonates to bidentate bicarbonates is pivotal to boost CO2 release, a process that is computed to be endergonic by ca. 1.90 eV. Given that this energy input is important, the greater abundance of the (100) facet on ceria crystals can be tentatively related to a poorer overall CO oxidation activity.
 +1 credit
+1 credit