Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oscar Campuzano | + 6924 word(s) | 6924 | 2022-01-10 07:53:51 | | | |
| 2 | Bruce Ren | Meta information modification | 6924 | 2022-01-12 10:05:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Campuzano, O. Clinical Genetics of Inherited Arrhythmogenic Disease in children. Encyclopedia. Available online: https://encyclopedia.pub/entry/18119 (accessed on 29 July 2026).
Campuzano O. Clinical Genetics of Inherited Arrhythmogenic Disease in children. Encyclopedia. Available at: https://encyclopedia.pub/entry/18119. Accessed July 29, 2026.
Campuzano, Oscar. "Clinical Genetics of Inherited Arrhythmogenic Disease in children" Encyclopedia, https://encyclopedia.pub/entry/18119 (accessed July 29, 2026).
Campuzano, O. (2022, January 12). Clinical Genetics of Inherited Arrhythmogenic Disease in children. In Encyclopedia. https://encyclopedia.pub/entry/18119
Campuzano, Oscar. "Clinical Genetics of Inherited Arrhythmogenic Disease in children." Encyclopedia. Web. 12 January, 2022.
Copy Citation
Sudden death is a rare event in the pediatric population but with a social shock due to its presentation as the first symptom in previously healthy children. Comprehensive autopsy in pediatric cases identify an inconclusive cause in 40–50% of cases. In such cases, a diagnosis of sudden arrhythmic death syndrome is suggested as the main potential cause of death. Molecular autopsy identifies nearly 30% of cases under 16 years of age carrying a pathogenic/potentially pathogenic alteration in genes associated with any inherited arrhythmogenic disease. In the last few years, despite the increasing rate of post-mortem genetic diagnosis, many families still remain without a conclusive genetic cause of the unexpected death.
Brugada syndrome
catecholaminergic polymorphic ventricular tachycardia
channelopathies
long QT syndrome
short QT syndrome
1. Introduction
While sudden cardiac death (SCD) is a rare event in pediatrics, it has a significant social impact, since it often presents as the first symptom in previously healthy children. The reported incidence rate of SCD in children and young adults is estimated to be between 1.3 and 1.7 per 100,000 persons-year [1][2], with twice as many cases in males than females. SCD is almost 10 times higher in young adults aged 31–35 years, while very low in children aged 6–10 years of age [3]. In addition to age and sex, other demographic factors such as ethnicity, exercise habits and geographic location impact on the incidence and survival rate of SCD, but the causes remain to be established [4][5]. During the first year of life, unexpected deaths remain unexplained after a comprehensive autopsy accounts for the highest infant mortality rate and is usually labelled as sudden infant death syndrome (SIDS) [6].
The most prevalent etiologies identified by medico-legal autopsy in children and young adults are cardiomyopathies, accounting for about 30% of SCD [7]. Nevertheless, a negative autopsy result is the most common finding in pediatrics, accounting for 40–50% of cases in the population under 16 years old. This outcome suggests a diagnosis of sudden arrhythmic death syndrome (SADS), the leading cause of SCD in children [8]. In SADS, the regular heart rhythm is abruptly replaced by a lethal ventricular arrhythmia, in the context of a structurally normal heart [9], the so-called inherited arrhythmia syndromes (IASs) or cardiac channelopathies.
Thanks to recent technological advances in the field of genetics, a considerable number of potentially IASs-causing genes have been identified although not comprehensively characterized. Current panels used in genetic testing of IASs range from the most common genes recommended by the clinical guidelines to the less common genes described in a few families, in large part not definitively associated with any IASs. Those that can be tested in a fast and cost-effective approach in clinical practice remain a current matter of argument. Therefore, studying a larger number of genes represents new challenges, such as limitations in establishing valid associations between genes and phenotypes. To address this problem, in 2013 the National Institute of Health (NIH) encouraged the development of ClinGen (Clinical Genome Resource https://clinicalgenome.org/ accessed on 3 December 2021), an international consortium of geneticists, genomic scientists and experts in the clinical field, which has established an evidence-based gene curation approach to establish gene–disease associations. Recently, evaluations of IAS-associated genes according to ClinGen’s approach have been published [10][11][12][13]. A further major challenge is due to the large number of variants of uncertain significance (VUS) yielded in next-generation sequencing (NGS) studies. The American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines are the current gold standard for classifying genetic variants. However, these guidelines were designed mainly for the classification of genetic variants in recessive or dominant pathologies with complete penetrance. IASs are characterized as diseases with an autosomal dominant inheritance pattern (AD), incomplete penetrance and variable expressivity, thus some criteria of the ACMG/AMP guidelines present limitations for their classification. Together with its strict criteria, this leads to high rates of VUS in the genetic diagnosis of IASs [14][15][16][17], making it necessary to work with adjusted ACMG/AMP criteria for each pathology and gene. Some researchers in the field of primary arrhythmias have aimed to perform a quantitative implementation of the ACMG/AMP guidelines for IAS’s genetic testing, with success in reducing the VUS rate [18].
We provide a state of the art overview of the genetic diagnosis of IASs in the pediatric population and its translation into clinical practice, based on international expert guidelines, recent advances in evidence-based genetic curation, and IAS-focused variant classification. We will emphasize genotype–phenotype and variant–phenotype correlation. Our goal is to facilitate translation of the genetic diagnosis to the clinical area, helping in risk stratification, treatment and the genetic counseling of families.
2. Inherited Arrhythmia Syndromes
Cardiac channelopathies are caused by defects in the genes encoding sodium (Na+), potassium (K+) and calcium (Ca2+) ion channels or their associated proteins. Defects in these proteins impair the generation and transmission of the action potential (AP) predisposing the patient to fatal arrhythmias [19]. The arrhythmias are usually of the type ventricular tachycardia (VT) or rapid ventricular fibrillation (VF), and generally polymorphic [20]. Despite the fact that each channelopathy usually has a characteristic electrocardiogram (ECG) profile, an associated clinical phenotype and specific genes involved, these factors can often be shared by two or more syndromes, leading to difficulties in establishing a differential diagnosis. In addition, some of these syndromes are not associated with baseline ECG abnormalities, which make them difficult to diagnose, with the added distress that SCD or resuscitated cardiac arrest is the initial symptom in most cases [21]. Therefore, an accurate genetic diagnosis can help to predict the prognosis and management of patients and their families. IASs are predominantly monogenic syndromes with an AD inheritance pattern, characterized by incomplete penetrance and variable expressivity [22]. Phenotypes with an autosomal recessive (AR) and X-linked inheritance pattern occur, but they are a minority [23]. Four major IASs can be typically observed in pediatrics: long QT syndrome (LQTS), short QT syndrome (SQTS), Brugada syndrome (BrS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) [24].
3. Long QT Syndrome
LQTS is the most common of the IASs and the main contributor for SCD in young people under 20 years of age. LQTS affects approximately 1 per 2000–2500 persons-year [25] with a slight predominance of females [26]. Moreover, it is the major arrhythmogenic syndrome, responsible for infants’ deaths, especially in the first days of life [27]. It is characterized by QT interval prolongation and T-wave abnormalities in ECG, polymorphic ventricular tachycardia (PVT) in torsade de pointes (TdP), VF and syncope or SCD [28]. LQTS can be hereditary, congenital or acquired, usually associated with drugs and electrolyte imbalance (hypokalemia) (http://www.torsades.org accessed on 3 December 2021).
3.1. Genetics
Originally characterized as an AD inheritance syndrome, called Romano–Ward syndrome (LQT1–6 and LQT9–13), today 17 LQTS-associated genes are known (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, SCN1B, SCN4B, SCN5A, SNTA1 and TRDN) (Table 1) [29][30]. Most manifest with an AD inheritance pattern, except for the Jervell and Lange–Nielsen syndromes (JLNS) [31] and the recently characterized triadin knock out syndrome (LQT17) [32], which are inherited in an AR manner. In the most recent evaluation by an expert consensus, only three of these genes were considered to have adequate evidence to be classified as definitive for typical LQTS (KCNQ1, KCNH2, SCN5A) and four genes for LQTS with atypical features (CALM1, CALM2, CALM3, TRDN). The remaining 10 genes, despite being associated with LQTS, were considered to require further evidence for their classification as causals [10].
Table 1. Genotype-phenotype IASs correlation. Based on ClinGen [13]. Adapted from Nakajima, et al. [33]. ACM: Arrhythmogenic cardiomyopathy; AE3: Anion exchanger; AF: Atrial fibrillation; AFL: Atrial flutter; AR: autosomal recessive; ATS: Andersen-Tawil syndrome; AVB: atrioventricular block; BrS: Brugada syndrome; CDSP: Systemic primary carnitine deficiency; CPVT: catecholaminergic polymorphic ventricular tachycardia; CND: complex neurodevelopmental disorder; CRDS: Ca2+-release deficiency syndrome; DCM: Dilated cardiomyopathy; DD: developmental delay; DM: diabetes mellitus; ERS: Early repolarization syndrome; HCM: cardiomyopathy; Hypertrophic GOF: gain-of-function; ICa: Voltage-gated calcium currents; Ih: Hyperpolarization-activated non-selective cation channels/currents; IK: Delayed rectifier potassium currents; IK-Ach: Acetylcholine-activated inward rectifier potassium currents; IK-ATP: ATP-sensitive inward rectifier potassium currents; IKr: Rapidly activating IK; IKs: Slowly activating IK; IK1: Inward rectifier potassium currents; INa: Voltage-gated sodium currents; Ito: Transient outward potassium currents; TKOS: triadin knockout syndrome, TS: Timothy syndrome; JLNS: Jervell and Lange–Nielsen syndrome; LOF: loss-of-function; LQTS: Long QT syndrome; LVNC: Left ventricular non-compaction; PCCD: progressive cardiac conduction disease; SCA: Spinocerebellar ataxia; SND: Sinus node dysfunction; SNHL: Sensorineural hearing loss; SQTS: Short QT syndrome; SVT: Supraventricular tachyarrhythmia; WPW: Wolff–Parkinson–White.; XD: X-linked dominant.
| Cardiac Phenotype | Inheritance Model | Frequency | Gene Curation |
Genes | Main Type of Mutations |
Current Affected |
Non-Cardiac Phenotype |
Phenotypic Overlap (Both LOF/GOF Variants) |
Ref. |
|---|---|---|---|---|---|---|---|---|---|
| LQTS | |||||||||
| LQT1 | AD | 30–35% | Definitive genes | KCNQ1 | LOF | IKs | SNHL (AR), seizures |
JLNS (AR), SQTS, AF | [10][33][34][35][36][37][38] |
| LQT2 | AD | 25–30% | KCNH2 | LOF | IKr | Seizures | SQTS, BrS, AF | [10][33][34][39] | |
| LQT3 | AD | 5–10% | SCN5A | GOF | INa | Multiple (including seizures) |
BrS, SQTS, CPVT, ERS, AF, AFL, ARVC/D, HCM, DCM, LVNC, SVT, AVB, SND, PCCD, WPW | [10][33][34][40][41][42] | |
| LQT14–16 | AD | <1% | Definitive genes with atypical characteristics | CALM1–3 | GOF | ICa | Seizures, DD | CPVT | [10][33][43][44][45] |
| LQT17 (TKOS) | AR | <1% | TRDN | LOF | ICa | Muscle weakness | CPVT | [10][33][46][47] | |
| LQT5 | AD | <1% | Genes with moderate or limited evidence, associated with multiorgan syndromes |
KCNE1 | LOF | IKs | SNHL (AR) | JLNS (AR) | [10][33][34][48] |
| LQT8 | AD | <1% | CACNA1C | GOF | ICa | Dysmorphic and neurodevelopmental features | TS, SQTS, BrS, ERS, HCM, AF, SND, CND | [10][33][49][50][51][52][53] | |
| LQT7 | AD | <1% | KCNJ2 | LOF | IK1 | Muscle weakness, dysmorphic features, DD, seizures |
ATS, SQTS, AF, CPVT, DCM |
[10][33][37][42][54][55][56][57][58] | |
| LQTS | AD | <1% each | Other genes with limited evidence | ANK2 KCNJ5 KCNE2 AKAP9 SCN4B CAV3 SNTA1 |
LOF LOF LOF LOF GOF GOF GOF |
Many IK-Ach IKr IKs INa INa INa |
Seizures | CPVT, BrS, CND AF, SND AF |
[10][33][34][42][48][59][60][61][62][63] |
| BrS | |||||||||
| BrS1 | AD | 20–30% | Definitive gene | SCN5A | LOF | INa | Multiple (including seizures) |
BrS, SQTS, CPVT, ERS, AF, AFL, ARVC, HCM, DCM, LVNC, SVT, AVB, SND, PCCD, WPW | [12][64][65][66][67][68][69][70][71][72][73][74][75][76] |
| BrS | AD | <5% | Genes with moderate or limited evidence |
ABCC9 ANK2 KCNH2 KCNJ8 KCND3 KCNE3 CACNA1C CACNB2 CACNA2D1 HCN4 PKP2 GPD1-L TRPM4 SCN1B–3B SCN10A SLMAP RANGRF |
GOF GOF GOF GOF GOF GOF LOF LOF LOF LOF LOF LOF LOF LOF LOF LOF LOF |
IK-ATP Many IKr IK-ATP Ito Ito ICa ICa ICa Ih INa INa INa INa INa INa INa |
Seizures Seizures Seizures, SCA (See LQT8) |
ERS, AF, DCM LQTS, CPVT, CND LQTS, SQTS, AF ERS, AF ERS, AF, CND AF Multiple (see LQT8) SQTS, ERS, CND SQTS, ERS, CND AF, SND ARVC, DCM, ACM, CPVT DM LQTS AF AF |
[12][33][37][64][77][78][79][80][81] |
| XD | KCNE5 | LOF | Ito | AF | |||||
| CPVT | |||||||||
| CPVT1 | AD | 55–60% | Definitive genes | RYR2 | GOF | ICa | LQTS, HCM, LVNC, CRDS | [11][48][82][83][84][85][86][87] | |
| CPVT2 | AR | 3–5% | CASQ2 | LOF | ICa | [11][48][88] | |||
| CPVT3 | AR | 1–2% | TECRL | LOF | ICa | [11][48][89][90] | |||
| CPVT4 | AD | <1% | CALM1–3 | LOF | ICa | Seizures, DD | LQTS | [11][43][44][45][48] | |
| CPVT5 | AR | 1–2% | TRDN | LOF | ICa | Muscle weakness | LQTS | [11][47][48][91] | |
| CPVT | AD | <1% | Genes with moderate or limited evidence |
SCN5A PKP2 ANK2 KCNJ2 |
LOF LOF LOF LOF |
INa INa Many IK1 |
Multiple Seizures Seizures (See LQT7) |
Multiple (see BrS1) ARVC, DCM, ACM, CPVT LQTS, BrS, CND Multiple (see LQT7) |
[11][48][92] |
| SQTS | |||||||||
| SQT1 | AD | 15% | Definitive gene | KCNH2 | GOF | IKr | Seizures | LQTS, AF, BrS | [11][33][34][93] |
| SQT2–3 | AD | <5% each | Genes with strong-moderate evidence | KCNQ1 KCNJ2 |
GOF GOF |
IKs IK1 |
(See LQT1) (See LQT7) |
JLNS (AR), SQTS, AF Multiple (see LQT7) |
[10][11][33][34][35][36][37][38] |
| SQTS | AD | <1% | Gene with moderate evidence |
SLC4A3 | LOF | AE3 | [11][94] | ||
| SQTS | AD AR |
<1% each | Genes with limited evidence |
CACNA1C CACNB2 CACNA2D1 SCN5A SLC22A5 |
LOF LOF LOF LOF LOF |
ICa ICa ICa INa INa |
(See LQT8) Multiple Metabolic decompensation, skeletal myopathy |
Multiple (see LQT8) SQTS, ERS, CND SQTS, ERS, CND Multiple (see BrS1) CDSP |
[11][33][65][95][96][97] |
3.2. Definitive Genes for LQTS
Pathogenic variants in 3 genes (KCNQ1, KCNH2 and SCN5A) are responsible for approximately 90% of all diagnosed cases of LQTS [98]. With LQT1 (KCNQ1) accounting for 30–35% of cases, LQT2 (KCNH2) between 25–30% and LQT3 (SCN5A) around 5–10% [99][100]. The age of onset of clinical manifestations in each type is variable. LQT1 affect predominantly the pediatric population between 5–15 years of age, whereas LQT2 and LQT3 occur during puberty or later [34][101]. Even though severe causes of neonatal or even fetal manifestation have been reported, a similar mechanism for arrhythmogenesis is found in types 1 and 2. Both syndromes are caused by loss-of-function pathogenic variants in genes encoding for K+ channels. Disruption of these channels causes a repolarization delay with an increase in the AP (phase 3), leading to the QT interval prolongation. In comparison, in LQT3, this phenotype is caused by gain-of-function pathogenic variants in the SCN5A gene, which codes for a Na+ channel [102]. Adrenergic stimuli have an important role as a trigger of symptoms in LQTS types 1 and 2. Physical exercise is the main trigger of SCD in LQT1. A strong sudden startle, loud noise or emotional stress are triggers of LQT2. Moreover, LQT3 presents with malignant arrhythmias at rest or during sleep [34].
3.3. Definitive Genes for LQTS with Atypical Characteristics
Loss-of-function pathogenic variants in the CALM1, CALM2, CALM3 and TRDN genes cause LQTS types 14 to 17, respectively. The CALM, CALM2 and CALM3 genes code for calmodulin, and the TRDN gene for triadin, proteins involved in calcium-dependent processes and ion channel regulation [43][46]. LQT14–16 present atypical features, including seizures and neurodevelopmental delay [103] with symptoms manifesting in infants and young children between the ages of 0–5 years old and a high mortality rate [104]. LQT17 or triadin knockout syndrome (TKOS) also has poor prognosis, and exercise-induced SCD occurs in children aged 0–3 years old. Almost all patients are symptomatic by the age of 10 years old. The observation of negative T waves in the precordial leads is characteristic of LQT17 [47][105].
3.4. Genes with Moderate or Limited Evidence for LQTS
According to the recent evaluation by Adler et al., there is insufficient evidence to classify 10 of the 17 LQTS-related genes (CACNA1C, AKAP9, ANK2, CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4 and SNTA1) as LQTS-causing genes [31983240]. Each of these types represents less than 1% of LQTS. Some of them are associated with phenotypic syndromes and specific ECG features (Table 1). These include Andersen–Tawil syndrome (LQT7, KCNJ2) in which skeletal developmental abnormalities are observed [54]; Timothy syndrome (LQT8, CACNA1C) with characteristic neurological, facial and limb features [106] and JLNS syndrome, associated with sensorineural deafness (KCNQ1-KCNE1 genes) [107]. Although the association of pathogenic variants in these genes with multiorganic syndromes is clear, the level of evidence for the specific cardiac phenotype is not so clear, and further studies are needed [10].
3.5. Genetic Modifiers and Acquired LQTS
GWAS studies have identified polymorphisms associated with increased risk of triggering LQTS [108][109]. Common variants in the NOS1AP (Nitric Oxide Synthase 1 Adaptor Protein) gene confer an increased risk of SCD in patients with LQT1 [110]. Likewise, some variants have a protective effect, as is the case of p.H558R (SCN5A), which reduces the pathogenic effect of other pathogenic variants, producing a less severe phenotype [22]. The common variants p.D85N (KCNE1) and T8A-MiRP1 (KCNE2) in Caucasians [111][112] and p.S1103Y (SCN5A) in African Americans [113], confer risk in the presence of other triggers such as drugs, a phenotype known as acquired LQTS (aLQTS). These variants are insufficient by themselves to cause LQTS in the absence of other interval-prolonging factors [10][114]. However, they are of major importance due to the high frequency of aLQTS [115].
3.6. Diagnosis
According to the 2015 ESC Guideline, LQTS is diagnosed when the patient presents one of the following criteria: a corrected QT (QTc) ≥480 ms (repeated 12-lead ECGs), a LQTS risk score >3, or when a pathogenic alteration in one of the LQTS-causing genes is identified [116]. The LQTS risk score combines the altered ECG parameters with the patient’s clinical and family history and is based on the clinical score proposed by Schwartz in 1993 [117]. Regardless, in the presence of unexplained syncope a QTc ≥460 ms is sufficient to diagnose LQTS [116] [26318695]. LQTS presents cardiac and extracardiac phenotypic features, as well as ECG characteristics that allow its classification, although 25% of individuals with positive genetics show a normal baseline ECG [118].
3.7. Risk Stratification
Multiple factors are known to raise the likelihood of SCD in patients with LQTS. The presence of a QTc >500 ms is the strongest of these predictors [119][120]. In children, this parameter can be modified by the patient’s age, sex and genotype, with a critical transition period between 12–14 years of age [121]. Patients who have suffered syncope during childhood have an increased risk of recurrent episodes, which can be reduced through the use of beta-blockers (BB) and/or implantable cardiac defibrillators (ICD) [122][123]. Among the types, LQT3 has a worse prognosis and the first presentation is usually SCD [119][124]. Likewise, LQT14–16 types have been associated with a very severe phenotype in infants and poor response to available therapies [103]. Women with LQT2 have an increased risk of SCD in the first 6 months postpartum, suggesting a potential hormonal effect [125]. Pathogenic variants type and location, as well as other additive genetic factors may increase the risk of SCD. For example, female adults with LQT2 run a higher risk of SCD than males. However, when missense pathogenic variants in the KCNH2 gene are in the pore loop regions, males are at a higher risk of SCD than their female counterparts [126]. Indeed, pathogenic variants in these regions have been found to be associated with QT interval prolongation and the development of TdP during fever, suggesting that fever may be a potential trigger of arrhythmias in patients with LQT2 [127].
3.8. Genetic Counselling
LQTS has incomplete penetrance and variable expressivity, even in the same family. The penetrance is estimated to be about 40%, a range that can vary depending on genotype, pathogenic variants type and location, age and sex, among other things [22][128]. The identification of a pathogenic variant in one of the LQTS-associated genes (Table 1) is important to establish a differential diagnosis of patients and their relatives. In LQTS, treatment and risk stratification differ depending on the gene causing the disease, and can be variant-specific [129]. According to guideline recommendations, only genes with definitive evidence for LQTS (KCNQ1, KCNH2 and SCN5A) should be routinely used in the evaluation of patients and their families. In patients with clinical findings consistent with the phenotypic expression demonstrated in LQTS with atypical features, related genes (CALM1, CALM2, CALM3 and TRDN) should also be tested [10]. Genetic testing can identify an LQTS-causing alteration in 70–80% of cases [99]. The proportion of LQTS caused by de novo pathogenic variants is difficult to estimate, but is expected to be low [130]. Between 5–9% of familial cases of LQTS have two or more pathogenic variants (biallelic or digenic), which have been associated with a more severe phenotype [131][132]. The coexistence of two or more pathogenic variants could explain the variable expressivity observed in some families [133]. The copy number variants (CNV) detection rate among LQTS families is not very clear, but is estimated to be between 2–11% [134][135][136][137][138]. Population screening by ECG has been promoted to identify individuals at risk of LQTS, which has been successful in reducing SCD rates among patient family members [139] neonates [27][140] and athletes [141][142].
3.9. Management and Treatment
In children with LQTS the first approach is to avoid genotype-specific triggers, such as competitive sports, especially swimming in LQT1, and exposure to loud noise in children with LQT2 as well as the avoidance of the QT interval prolonging drugs in all carriers of LQTS-associated variants (http://crediblemeds.org/ accessed on 3 December 2021) [116]. Long-acting BB (nadolol) are recommended in all types, including asymptomatic genetic carriers [143][144], as their use decreases the risk of SCD [145]. Specific treatment of LQT3 with mexiletine and/or flecainide has proven to be highly effective. In LQT1 the use of BB is very effective, and some authors suggest that it is not necessary to place an ICD in patients at low risk of SCD (asymptomatic prepubertal girls and adults >20 years with normal ECG) [146]. Left-cardiac sympathetic denervation is indicated in patients with LQT1 or when BB therapy is contraindicated or badly tolerated [147]. ICDs are used in patients at high risk of SCD despite previous therapies, in those who have previously presented syncope while taking BB, and effective LCSD (left cardiac sympathetic denervation) has been performed [148]. Despite its efficacy, ICD has a high economic cost and can present numerous complications. Its use in the pediatric population should be assessed on a case-by-case basis by specialists [149].
4. Brugada Syndrome
BrS in children and young adults is rare, and its incidence rate and clinical implications remain unclear. In the general population, its prevalence is estimated to be between 1 in 2000–5000 person-years [150]. The syndrome has a higher prevalence in Southeast Asian countries [151], and is more frequent among males than females [152]. It is characterized by a right bundle branch block, a very sharp T wave and spontaneous or drug-induced ST-segment elevation (J point) in the right precordial leads (V1–V3), known as ‘type-1’ BrS ECG pattern [64]. Clinical manifestations may appear between the ages of 2 months and 77 years old, but the mean age of presentation is 40 years old [153]. Symptoms present at rest, during sleep or febrile episodes, including nocturnal agonal respirations, palpitations, seizures, and polymorphic ventricular tachycardia (PVT) or VF. Most individuals remain asymptomatic, although SCD occurs in 17–42% of the cases and can be the initial presentation [154].
4.1. Genetics
Genetically described as a Mendelian syndrome with an autosomal dominant inheritance pattern and incomplete penetrance [155], recent evidence suggests that BrS may be an oligogenic disease, involving several genetic factors [156]. However, the lack of conclusive data on these genetic alterations leads it to remain classified as a monogenic syndrome [157]. To date, more than 20 genes have been associated with BrS (ABCC9, ANK2, CACNA1C, CACNA2D1, CACNB2, GPD1-L, HCN4, KCND3, KCNE3, KCNE5, KCNH2, KCNJ8, PKP2, RANGRF, SCN10A, SCN1B, SCN5A, SCN2B, SLMAP and TRPM4), reappraisal of these genes by Hosseini et al., established that only SCN5A had definitive evidence of being a causal gene [12] and its genetic analysis is the only one recommended by current guidelines [23][158].
4.2. Definitive Gene for BrS
Loss-of-function pathogenic variants in the SCN5A gene account for approximately 30% of genetically positive BrS cases. This gene encodes the alpha subunit of the cardiac sodium channel Nav1.5, responsible for phase 0 of the AP. Inactivation of the channel leads to a delay in ventricular polarization, resulting in the development of ventricular tachycardia and fibrillation (VT/VF) [150].
4.3. BrS2–12 and Other Susceptibility Genes with Limited Evidence
Pathogenic variants associated with BrS2–12, genes (GPD1-L, CACNA1C, CACNB2, SCN1B, KCNE3, SCN3B, HCN4, KCND3, KCNJ8, CACNA2D1 and MOG1) together represent less than 5% of all diagnosed cases [159]. Over the past few years, other genes have been suggested as possible causes of BrS (ABCC9, ANK2, FGF12, HEY2, KCND2, KCNH2, KCNE5, LRRC10, SEMA3A, PKP2, RANGRF, SCN10A, SCN2B, SLMAP and TRPM4), but no comprehensive clinical and cellular studies have confirmed this association [157]. All of them follow an AD inheritance pattern, except for the KCNE5 gene, which follows an X-linked dominant pattern [160][161].
4.4. Diagnosis
According to the 2015 ESC guidelines, BrS is diagnosed in patients with a ‘type 1’ ECG pattern, ST-segment elevation ≥2 mm (J-point) in one or more of the right precordial leads (V1–V3) [116]. This ECG pattern may occur spontaneously or be unmasked by a provocation test with a class Ic drug (sodium channel blockers such as ajmaline, flecainide, procainamide, or pilsicainide), in this case additional clinical criteria are required for diagnosis [64]. Fever is a trigger for ventricular arrhythmias in patients with BrS and may unmask the characteristic ECG pattern, especially in children under 5 years of age [162]. A 12-lead ECG is recommended during febrile episodes in children with a family history of BrS and in all children with febrile seizures [163]. Initially presumed not to have any structural abnormalities, postmortem histological studies and endomyocardial biopsies have shown changes at the tissular and molecular level in patients with BrS. These changes include localized electroanatomical and structural abnormalities in the right ventricular outflow tract (RVOT), fibrosis, fatty infiltration, increased epicardial collagen, and decreased expression of Connexin 43 at right ventricular gap junctions [164][165][166].
4.5. Risk Stratification
Despite advances in risk stratification of IAS, in BrS it remains challenging. The most important risk marker is the presentation of a previous arrhythmogenic event (AE, AF, syncope, or SCD), which increases the likelihood of SCD in both young and adults [167][168]. The age of onset is a notable prognostic marker. For instance, although BrS is uncommon in children, they present with a more severe form of the disease [169]. Gender is an important risk factor, with males being up to 5–8 times more affected than females, presenting a more severe phenotype, earlier symptom debut and a higher number of events [152][170]. These gender differences have not been observed in children under 12 years old [167]. Family history of SCD and the presence of pathogenic variants in SCN5A could also be predictors of high risk in adults and adolescents, [167] even though their role in risk stratification is still controversial [171][172]. Other risk factors observed are spontaneous variation of the ‘type 1’ ECG pattern and fragmentation of the QRS complex [171][173], nevertheless further studies are required to confirm this association.
4.6. Management and Treatment
In children with BrS or a family history, BrS-inducing drugs should be avoided (http://www.brugadadrugs.org accessed on 3 December 2021). Appropriate treatment of any fever with antipyretic drugs should be provided [167]. ICD implantation is the only treatment that reduces the risk of SCD in BrS. It is indicated in all patients resuscitated from arrhythmic syncope, those with documented VT or VF or with a spontaneous ‘type 1’ ECG pattern [116][149]. In patients with electrical storms, administration of isoproterenol is recommended [174][175]. Quinidine is recommended for patients who refuse ICD implantation or in those who, despite having an ICD, still have a high risk of SCD [176][177]. Catheter ablation is useful in high-risk patients with a history of electrical storms or repeated appropriate ICD shocks [178]. ICD implantation in asymptomatic individuals with a ‘type 1’ ECG pattern for primary prevention, including children, remains controversial and is a challenge for specialists who have to handle it on a case-by-case basis [149][177].
4.7. Genetic Counseling
BrS penetrance is highly variable in the different published studies. It is estimated to range from 12.5% to 50% [22][179]. About 70–80% of families with BrS do not have a genetic diagnosis. However, although the results of genetic screening do not currently influence prognosis or treatment, genetic testing should be performed in all first-degree relatives if the index case tested positive [99][116]. Moreover, the ECG pattern of BrS ‘type 1’ is uncommon in children and genetic testing may help with their diagnosis [21]. In BrS families counseling should also include an ECG, because negative-genotype positive-phenotype cases are not uncommon [180]. The proportion of cases caused by de novo pathogenic variants is estimated at 1% and the number of cases with a CNV variant is approximately 1.3–2.9% [138][181]. ECG screening using a provocation test for BrS detection is controversial [182][183], but should be performed when an abnormal ECG is found. ECG screening has benefits in the prevention of SCD in neonates [140] and the young population [184].
5. Short QT Syndrome
SQTS is an extremely rare inherited disease associated with SCD. To date, less than 200 cases have been reported worldwide [93]. The estimated prevalence varies between 0.18–2.9%, with a higher incidence in males than females [25]. The incidence rate can even be lower (0.02–0.10%) if more restrictive values are considered for its diagnosis [185][186]. While prevalence of the syndrome in children and adolescents is low (about 0.05%), early detection is important, as it is potentially lethal for all age groups. It is characterized by a short QT interval on the ECG (<330 ms), with an asymmetric and peaked T wave. Symptoms occur mostly in men between the ages of 14 and 40 and may be favored by hormonal causes [187]. Cardiac events usually occur in adrenergic situations (noise or exercise), although it can also occur at rest. Clinical presentation includes ventricular repolarization abnormalities (AF and VT) and syncope. The probability of presenting SCD as the first symptom increases with age, reaching 41% at the age of 40 [188]. Currently, approximately 40% of cases remain asymptomatic [189].
5.1. Genetics
At present, nine genes have been associated with SQTS (CACNA1C, CACNA2D1, CACNB2, KCNH2, KCNJ2 and KCNQ1, SLC22A5, SLC4A3 and SCN5A) [190]. Evaluation of these genes, by Walsh et al., showed that only the KCNH2 gene had definitive evidence for SQTS causality. Three other genes (KCNQ1, KCNJ2, SLC4A3) presented strong to moderate evidence. Causality of the other SQTS-associated genes remains still in dispute [11]. These data are consistent with the findings published by Campuzano et al., who found that all variants with a conclusive pathogenic role in SQTS clustered in three genes (KCNQ1, KCNH2 and KCNJ2). In that study, the SLC4A3 gene was excluded, since carriers were in a gray zone of SQTS diagnosis (with a QTc ≤370 ms) [190].
5.2. SQT1 Definitive Gene: KCNH2
The p.T618I and p.N588K pathogenic variants in the KCNH2 gene are the most frequent associated with SQTS, accounting for 85% of SQT1 and 55% of all genetically identified cases of SQTS [189]. Gain-of-function pathogenic variants in KCNH2 lead to prolonged K+ channel activation and accelerated cardiac repolarization with shorter refractory periods, potentially triggering life-threatening supraventricular and ventricular arrhythmias [189].
5.3. Genes with Strong or Moderate Evidence for SQTS
SQT2 and SQT3 are driven by gain-of-function pathogenic variants in genes encoding for K+ channels (KCNQ1 and KCNJ2, respectively). The mechanism of arrhythmogenicity is similar to that presented by KCNH2. The SLC4A3 gene, recently associated with SQTS, presents an uncommon mechanism for the development of malignant arrhythmia. SLC4A3 encodes the plasma membrane anion exchange protein 3 (AE3) and acts by mediating part of the Cl−/HCO3− exchange in cardiac myocytes. Loss-of-function pathogenic variants in the SLC4A3 gene would cause an increase in pHi and a decrease in [Cl−]i, shortening the duration of the AP [94].
5.4. Diagnosis
According to the 2015 ESC guidelines, SQTS is diagnosed by the presence of a QTc ≤340 ms, or ≤360 ms when one the following clinical criteria occur: the detection of a known pathogenic alteration, a family history of SQTS, a family history of SCD before the age of 40 years, or reanimated cardiac arrest with a structurally normal heart [116].
5.5. Risk Stratification
On account of the limited number of patients with SQTS and the phenotypic variability of the syndrome, risk stratification currently represents a challenge. To date, the only predictor of SCD found in patients with SQTS is a history of cardiac arrest [93]. Genotype-phenotype correlation studies have found that SQTS1 manifests at an older age and patients have a shorter QTc than other patients with SQTS. Nevertheless, no association of this reduction with an increased risk of SCD has been found [191].
5.6. Management and Treatment
ICD implantation is recommended for all patients with SQTS, especially for patients who have survived an aborted cardiac arrest or have presented spontaneous sustained VT [186][192]. QT interval prolonging drugs (quinidine and sotalol) should be considered for all patients at risk of SQTS in both, asymptomatic and symptomatic patients who don’t have an ICD, especially young children [186].
5.7. Genetic Counselling
Due to the low number of cases, the penetrance of SQTS is difficult to estimate. However, pathogenic variants with a penetrance of 100% have been reported [189]. The diagnostic yield of genetic testing in SQTS is low (<25%) [186]. Current guidelines recommend analysis of five genes: KCNH2, KCNQ1, KCNJ2, KCNJ2, CACNA1C and CACNB2 in the diagnosis of SQTS [116], with the KCNH2 gene as the most cost-effective option [193]. Familial genetic analysis is recommended, both to clarify the pathogenic role of newly identified variants and to identify family members at risk for SCD. To date, there is no published data on CNV analysis on patients with SQTS. De novo variants in the KCNQ1 gene have been associated with a particular in utero phenotype with clinical diagnosis of AF with concomitant bradycardia and short QT interval [194][195]. Some researchers support the screening of SQTS in the pediatric population, given its high lethality and the benefits of early diagnosis in the prevention of SCD. These studies have shown that the diagnostic criteria for QTc should be adjusted in each population based on factors including sex and age, to avoid false positives [196][197][198][199].
6. Catecholaminergic Polymorphic Ventricular Tachycardia
The prevalence of CPVT is estimated to be 1 per 10,000 population. However, the real prevalence is uncertain, as it might be underestimated due to its high lethality at a young age and difficulty in diagnosis [25][200]. CPVT is characterized by a bidirectional polymorphic VT, triggered by an adrenergic stimulus mainly during exertion, extreme stress or emotion that can lead to syncope and SCD. Syncopal episodes are increasingly reported during “awake rest” possibly due to anxiety, stress, or other psychological stimuli unrelated to exertion [201]. The age of onset can range from infancy to the age of 30, although it is more common in children aged 7–10 years old [202]. By the age of 10, about 35% of patients are symptomatic, increasing to 72% by the age of 21 [203]. A younger age of debut is often accompanied by more severe phenotypes and increased risk of SCD [42].
6.1. Genetics
According to the recent evaluation by Walsh et al., seven genes were classified as causing of CPVT with definite to moderate evidence. Four of them present an AD inheritance pattern (RYR2, CALM1, CALM2, CALM3) and three AR inheritance (CASQ2, TRDN, TECRL). Three genes (KCNJ2, PKP2, SCN5A) were reported for phenotypes that were not representative of CPVT, while the reported variants in the ANK2 gene were considered too common in the population to be disease-causing (Table 1) [11].
6.2. Definitive Genes for CPVT
CPVT1 is the most prevalent variant, accounting for more than 60% of all genetically diagnosed cases of CPVT [204]. CPVT1 with a high incidence in children around 10 years of age [205]. Thus, it is caused by pathogenic variants in the RYR2 gene, which encodes for ryanodine receptor 2, responsible for calcium regulation in the cardiomyocyte. A majority of pathogenic variants in the RYR2 gene are gain-of-function, which promote an increased Ca2+ release from the sarcoplasmic reticulum of cardiomyocytes into the cytoplasm leading to late after-depolarizations [48]. The remaining types of CPVT represent about 10% of cases. Most are caused by loss-of-function pathogenic variants in genes encoding proteins involved in the storage and release of Ca2+ in the sarcoplasmic reticulum [206]. CPVT2 is caused by homozygous or compound heterozygous pathogenic variants in the CASQ2 gene, following an AR inheritance model. CPVT2 accounts for about 3–5% of cases [203], affecting mainly children around the age of 7 years [205]. Pathogenic variants in TRDN, CALM1 and TECRL are responsible for CPVT3, CPVT4, and CPVT5, respectively. Each represents about 1–2% of the cases. CPVT3 and CPVT5 follow an AR inheritance pattern, even though AD inheritance has also been observed in some families [42][91]. In contrast, CPVT5 is presented with an AD inheritance pattern, a younger age of onset (approximately 2.3 years) and more severe phenotypes, being highly lethal in children [205][207]. Along with TRDN and CALM1, gain-of-function pathogenic variants in the CALM2 and CALM3 genes have been associated with the development of atypical CPVT, presenting more complex and variable associated phenotypes than classic [208][209].
6.3. Diagnosis
CPVT is diagnosed when exercise- or emotion-induced bidirectional or polymorphic VT is detected, in the presence of a structurally normal heart or in patients carrying pathogenic alterations in the definitive genes for CPVT [200]. Since the resting ECG is usually normal, ECG during exercise and Holter monitoring play a relevant role in the diagnosis. The disease can be easily missed or misdiagnosed; for instance, many children are initially diagnosed with epilepsy, as syncope may be associated with seizure movements.
6.4. Risk Stratification
Between 30–50% of patients with CPVT will experience SCD before the age of 30 [210][211]. The event rate in untreated children under 8 years old has been estimated at 58% that can be reduced to 27% with adherence to BB treatments [203]. RYR2 is one of the most prevalent genes in cohorts of patients with unexplained SCD, occurring in 5–10% of cases [212]. While the variable expressivity of the CPVT phenotype could be explained by the influence of other genetic and non-genetic factors, no genetic modifiers have been identified in CPVT to date [108][213]. Patients with the recessive form of CPVT and a younger age at diagnosis have a more severe phenotype [48][203]. The location of the rare variant may be a possible disease modifier [201]. No gender- or age-dependent differences in arrhythmic risk in children have been found to date [214]. However, further studies in CPVT risk stratification are needed to draw definitive conclusions.
6.5. Management and Treatment
In children with CPVT, avoidance of phenotype triggers such as competitive sports, strenuous exercise (especially swimming), and stressful environments is recommended. BB are the first-line of treatment, their use is recommended in all patients, even in genetically identified asymptomatic patients [116]. About 25% of children experience syncope or cardiac arrest despite treatment with BB [215]. In these patients, it is advisable to include flecainide therapy and/or left cardiac sympathetic denervation (LCSD). LCSD has been proven to reduce the rate of arrhythmic events in patients with LQTS and CPVT [200][216]. It should be noted that ICD therapy may be counterproductive in CPVT, because the discharge may activate adrenergic production and exacerbate the VT storm, so its implantation should be assessed by the specialist [217].
6.6. Genetic Counseling
CPVT penetrance can vary between 63–78% [22][213][218]. The diagnostic yield is high, with a positive genetic result in 60–65% of the studied cases [25]. Genetic testing is recommended by expert consensus, with RYR2 and CASQ2 genes as the most cost-effective options [23]. Identification of family members at risk is critical to avoid SCD, which is the first manifestation in up to 30–50% of cases [210]. CPVT has a high incidence of de novo variants, which are found in approximately 50% of genetically diagnosed CPVT patients [25]. CNV in RYR2 have been associated with CPVT [138][219][220], while the other CPVT-related genes have not been examined so far.
7. Genetic Overlap
Several causative genes for IASs are common to two or more syndromes (LQTS, SQTS, BrS, and CPVT) (Figure 1) and have been found to overlap with other cardiac and extracardiac clinical phenotypes (Table 1). This genetic overlap could be explained since pathogenic variants can alter different properties of the channels or proteins, affecting ion exchange, interaction with auxiliary proteins, as well as gene expression. The final phenotype would depend not only on which property is affected, but also how it is affected. Notably, some genes that cause LQTS are the same genes that also cause SQTS (KCNQ1, KCNH2, KCNJ2, CACNA1C, CACNB2, and CACNA2D1), CPVT (CALM1, CALM2, CALM3, TRDN, KCNJ2, SCN5A, ANK2 and TECRL) or BrS (SCN5A) [42][89]. However, the pathogenic variants usually cause an opposite effect on the channel.
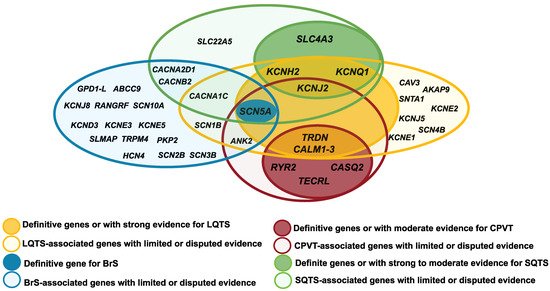
Figure 1. Diagram of the overlap between the genes IASs-associated genes: Brugada syndrome (BrS); short QT syndrome (SQTS); long short QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT).
Some pathogenic variants have been found to lead to complex overlapping forms of two or more syndromes. Several publications support the fact that pathogenic variants in KCNQ1, ANK2, KCNE1, KCNE2, KCNH2, KCNJ2 and SCN5A can lead to a complex overlapping phenotype of LQTS, CPVT and ventricular ectopy [42][48][92]. Hirose et al., recently described in a cohort of children (<16 years old) the association of loss-of-function pathogenic variants in RYR2 with various types of arrhythmia, including LQTS, VF and scTdP, depending on the alteration of channel activity [83][84]. The pathogenic variants p.I4855M and deletion of exon 3 in the RYR2 gene have been associated with the rare syndrome of left ventricular non-compaction (LVNC) overlap and CPVT, presenting a high lethality [85]. Pathogenic variants in the TRPM4 gene, both gain and loss of function, have been identified in patients with different forms of cardiac disorder including conduction defects, BrS and LQTS [77]. The PKP2 gene, the main gene mutated in arrhythmogenic cardiomyopathy (ACM), has been recently associated with BrS and CPVT. The p.S183N pathogenic variant has been reported in both a patient with BrS and a patient with a definite diagnosis of ACM [221]. However, further studies are needed to establish the pathophysiological mechanisms of these correlations.
7.1. SCN5A Clinical Overlap
Pathogenic variants affecting SCN5A have been found in all major IAS, as well as in other associated cardiac phenotypes (Figure 2). Gain-of-function pathogenic variants in SCN5A (LQT3), have been associated with other arrhythmias including multifocal ectopic Purkinje-related premature contractions [222][223] and atypical CPVT-like phenotype [224]. For instance, p.T1857I and p.I141V variants, have been associated with tachyarrhythmias and exercise-induced polymorphic ventricular arrhythmia [224][225]. However, the correlation between SCN5A pathogenic variants and CPVT remains debated. Meanwhile, loss-of-function pathogenic variants in SCN5A (usually associated with BrS) have also been implicated in certain phenotypes including isolated cardiac conduction defect and sick sinus syndrome (SSS) [226][227]. In addition, both loss-of-function and gain-of-function pathogenic variants can cause dilated cardiomyopathy (DCM), AF, and overlap syndromes [226][228]. The founder pathogenic variants p.E1784K [229][230], p.F1617del [231], p.1795insD [232], among others, can be manifested as a mixed clinical phenotype of LQTS and/or BrS, even between affected individuals in the same family. Recently, Sasaki, et al. have described the p.A735E pathogenic variant, which could be associated with the overlap of multiple phenotypes in eight carrier individuals, although further studies are needed to ratify this hypothesis [76].
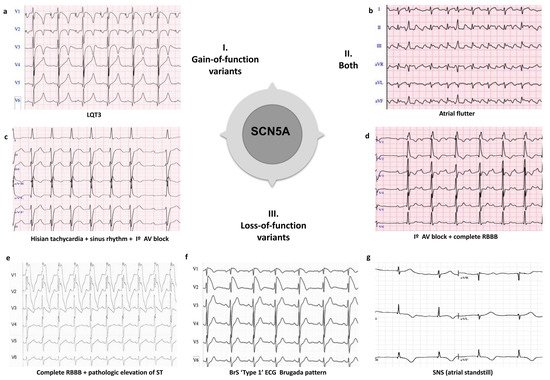
Figure 2. Examples of clinical overlap due to pathogenic variants in the SCN5A gene. I. Phenotype caused by gain-of-function variants in the SCN5A gene: (a) LQTS (Long QT syndrome type 3); II. Phenotype caused by both gain-of-function and loss-of-function variants in the SCN5A gene: (b) Atrial flutter; III Phenotypes caused by loss-of-function variants in the SCN5A gene: (c) Hisian tachycardia + sinus rhythm + I° AV block; (d) I° AV block + complete RBBB (right bundle branch block); (e) Complete RBBB + pathologic elevation of ST; (f) BrS (Brugada syndrome) ‘type 1’ ECG; and (g) SNS (sinus node syndrome), atrial standstill.
7.2. Genetic Overlap of Arrhythmogenic Phenotypes and Epilepsy
Recently, evidence for genetic overlap between IASs and epilepsy has been reported. Sudden unexpected death in epilepsy (SUDEP) share many features with SADS in the young and may have a similar genetic contribution [233][234]. In a systematic review, Anwar and Chahal, et al., found that 11% of the most frequent pathogenic variants identified by molecular autopsy in SUDEP were found in genes related to cardiac channelopathies [235].
7.3. Non-Genetic Phenotype Overlapping
Multiple phenotypic overlaps between IASs have been described, in which no underlying genetic cause has been observed ERS and BrS [66][236], BrS and CA [237], ACM and BrS [238]. The overlap of ACM and BrS is controversial, since their diagnostic criteria exclude the coexistence of both syndromes in the same individual. This overlap would not be fully explained by a genetic overlap. The ECG pattern of BrS (drug-induced) was observed both in patients with ACM and pathogenic variants in the SCN5A gene [239] as well as in patients without SCN5A pathogenic variants [240][241]. Therefore, it is possible that genetic (coding or non-coding variants) and non-genetic (demographic variables or exogenous factors) modifiers could be involved in this variability, all of them contributing additively to the expression of the phenotype. An alternative explanation for phenotypic overlap could be misdiagnosis due to the use of drugs in ECG testing. Notably, it has been shown that flecainide can induce ST-segment elevation in ECG in patients with LQTS3, and lead to misdiagnosis in some cases [242].
References
- Ha, F.J.; Han, H.-C.; Sanders, P.; Fendel, K.; Teh, A.W.; Kalman, J.M.; O’Donnell, D.; Leong, T.; Farouque, O.; Lim, H.S. Sudden Cardiac Death in the Young. Circ. Cardiovasc. Qual. Outcomes 2020, 13, e006470.
- Couper, K.; Putt, O.; Field, R.; Poole, K.; Bradlow, W.; Clarke, A.; Perkins, G.D.; Royle, P.; Yeung, J.; Taylor-Phillips, S. Incidence of sudden cardiac death in the young: A systematic review. BMJ Open 2020, 10, e040815.
- Bagnall, R.D.; Singer, E.S.; Tfelt-Hansen, J. Sudden Cardiac Death in the Young. Heart Lung Circ. 2020, 29, 498–504.
- Hayashi, M.; Shimizu, W.; Albert, C.M. The Spectrum of Epidemiology Underlying Sudden Cardiac Death. Circ. Res. 2015, 116, 1887–1906.
- Meyer, L.; Stubbs, B.; Fahrenbruch, C.; Maeda, C.; Harmon, K.; Eisenberg, M.; Drezner, J. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: A 30-year review. Circulation 2012, 126, 1363–1372.
- Goldberg, N.; Rodriguez-Prado, Y.; Tillery, R.; Chua, C. Sudden Infant Death Syndrome: A Review. Pediatr. Ann. 2018, 47, e118–e123.
- Morentin, B.; Suárez-Mier, M.P.; Monzó, A.; Molina, P.; Lucena, J.S. Sports-related sudden cardiac death due to myocardial diseases on a population from 1–35 years: A multicentre forensic study in Spain. Forensic Sci. Res. 2019, 4, 257–266.
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452.
- Tsuda, T.; Fitzgerald, K.K.; Templer, J. Sudden cardiac death in children and young adults without structural heart disease: A comprehensive review. Rev. Cardiovasc. Med. 2020, 21, 205–216.
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428.
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; Amenta, S.; Feilotter, H.; Nannenberg, E.A.; Mazzarotto, F.; et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur. Heart J. 2021, ehab687.
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of reported genes for sudden arrhythmic death: Evidence-based evaluation of gene validity for brugada syndrome. Circulation 2018, 138, 1195–1205.
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242.
- Denham, N.C.; Pearman, C.M.; Ding, W.Y.; Waktare, J.; Gupta, D.; Snowdon, R.; Hall, M.; Cooper, R.; Modi, S.; Todd, D.; et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2019, 30, 118–127.
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Coll, M.; Iglesias, A.; Ferrer-Costa, C.; Cesar, S.; Arbelo, E.; García-Álvarez, A.; Jordà, P.; et al. Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 2020, 54, 102732.
- Grassi, S.; Campuzano, O.; Coll, M.; Brión, M.; Arena, V.; Iglesias, A.; Carracedo, A.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Leg. Med. 2020, 45, 101712.
- Campuzano, O.; Sanchez-Molero, O.; Fernandez, A.; Mademont-Soler, I.; Coll, M.; Perez-Serra, A.; Mates, J.; del Olmo, B.; Pico, F.; Nogue-Navarro, L.; et al. Sudden Arrhythmic Death During Exercise: A Post-Mortem Genetic Analysis. Sports Med. 2017, 47, 2101–2115.
- Walsh, R.; Lahrouchi, N.; Tadros, R.; Kyndt, F.; Glinge, C.; Postema, P.G.; Amin, A.S.; Nannenberg, E.A.; Ware, J.S.; Whiffin, N.; et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genet. Med. 2021, 23, 47–58.
- Kline, J.; Costantini, O. Inherited Cardiac Arrhythmias and Channelopathies. Med. Clin. N. Am. 2019, 103, 809–820.
- Tse, G.; Chan, Y.W.F.; Keung, W.; Yan, B.P. Electrophysiological mechanisms of long and short QT syndromes. IJC Heart Vasc. 2017, 14, 8–13.
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Iglesias, A.; Arbelo, E.; Brugada, J.; Brugada, R. Genetics of inherited arrhythmias in pediatrics. Curr. Opin. Pediatr. 2015, 27, 665–674.
- Coll, M.; Pérez-Serra, A.; Mates, J.; Del Olmo, B.; Puigmulé, M.; Fernandez-Falgueras, A.; Iglesias, A.; Picó, F.; Lopez, L.; Brugada, R.; et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology 2017, 7, 3.
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339.
- Heying, R.; Albert, D.C.; Voges, I.; Sendzikaite, S.; Sarquella-Brugada, G.; Pluchinotta, F.; Brzezinska-Rajszys, G.; Stein, J.I.; Milanesi, O. Association for European Paediatric and Congenital Cardiology recommendations for basic training in paediatric and congenital cardiology 2020. Cardiol. Young 2020, 30, 1572–1587.
- Offerhaus, J.A.; Bezzina, C.R.; Wilde, A.A.M. Epidemiology of inherited arrhythmias. Nat. Rev. Cardiol. 2020, 17, 205–215.
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-qt syndrome. Circulation 2009, 120, 1761–1767.
- Sarquella-Brugada, G.; García-Algar, O.; Zambrano, M.D.; Fernández-Falgueres, A.; Sailer, S.; Cesar, S.; Sebastiani, G.; Martí-Almor, J.; Aurensanz, E.; Cruzalegui, J.C.; et al. Early Identification of Prolonged QT Interval for Prevention of Sudden Infant Death. Front. Pediatr. 2021, 9, 704580.
- Goldenberg, I.; Moss, A.J. Long QT Syndrome. J. Am. Coll. Cardiol. 2008, 51, 2291–2300.
- Waddell-Smith, K.E.; Skinner, J.R. Update on the Diagnosis and Management of Familial Long QT Syndrome. Heart Lung Circ. 2016, 25, 769–776.
- Skinner, J.R.; Winbo, A.; Abrams, D.; Vohra, J.; Wilde, A.A. Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ. 2019, 28, 22–30.
- Krishnan, M.N.; Pavithran, K. Jervell and Lange-Nielsen Syndrome. Available online: https://pubmed.ncbi.nlm.nih.gov/30725985/ (accessed on 31 October 2021).
- Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Cesar, S.; Arbelo, E.; Jordà, P.; García-Álvarez, A.; Cruzalegui, J.C.; Merchan, E.F.; Fiol, V.; Brugada, J.; et al. Pediatric Malignant Arrhythmias Caused by Rare Homozygous Genetic Variants in TRDN: A Comprehensive Interpretation. Front. Pediatr. 2021, 8, 754.
- Nakajima, T.; Tamura, S.; Kurabayashi, M.; Kaneko, Y. Towards Mutation-Specific Precision Medicine in Atypical Clinical Phenotypes of Inherited Arrhythmia Syndromes. Int. J. Mol. Sci. 2021, 22, 3930.
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT Syndrome: Genetics and Future Perspective. Pediatr. Cardiol. 2019, 40, 1419–1430.
- Neyroud, N.; Tesson, F.; Denjoy, I.; Leibovici, M.; Donger, C.; Barhanin, J.; Fauré, S.; Gary, F.; Coumel, P.; Petit, C.; et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 1997, 15, 186–189.
- Bellocq, C.; Van Ginneken, A.C.G.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.A.M.; Baró, I.; Wilde, A.A.M. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004, 109, 2394–2397.
- Feghaly, J.; Zakka, P.; London, B.; MacRae, C.A.; Refaat, M.M. Genetics of Atrial Fibrillation. J. Am. Heart Assoc. 2018, 7, e009884.
- González, A.; Aurlien, D.; Haugaa, K.H.; Taubøll, E. Epilepsy in patients with long QT syndrome type 1: A Norwegian family. Epilepsy Behav. Case Rep. 2018, 10, 118–121.
- Zamorano-León, J.J.; Yañez, R.; Jaime, G.; Rodriguez-Sierra, P.; Calatrava-Ledrado, L.; Alvarez-Granada, R.R.; Mateos-Cáceres, P.J.; MacAya, C.; López-Farré, A.J. KCNH2 gene mutation: A potential link between epilepsy and long QT-2 syndrome. J. Neurogenet. 2012, 26, 382–386.
- McNair, W.P.; Ku, L.; Taylor, M.R.G.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004, 110, 2163–2167.
- García-Cisneros, S.; Sánchez-Alemán, M.; Conde-Glez, C.J.; Lara-Zaragoza, S.J.; Herrera-Ortiz, A.; Plett-Torres, T.; Olamendi-Portugal, M. Performance of ELISA and Western blot to detect antibodies against HSV-2 using dried blood spots. J. Infect. Public Health 2019, 12, 224–228.
- Kallas, D.; Lamba, A.; Roston, T.M.; Arslanova, A.; Franciosi, S.; Tibbits, G.F.; Sanatani, S. Pediatric Catecholaminergic Polymorphic Ventricular Tachycardia: A Translational Perspective for the Clinician-Scientist. Int. J. Mol. Sci. 2021, 22, 9293.
- Badone, B.; Ronchi, C.; Kotta, M.-C.; Sala, L.; Ghidoni, A.; Crotti, L.; Zaza, A. Calmodulinopathy: Functional Effects of CALM Mutations and Their Relationship With Clinical Phenotypes. Front. Cardiovasc. Med. 2018, 5, 5.
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474.
- Reed, G.J.; Boczek, N.J.; Etheridge, S.P.; Ackerman, M.J. CALM3 mutation associated with long QT syndrome. Hear. Rhythm 2015, 12, 419–422.
- Rabbani, B.; Khorgami, M.; Dalili, M.; Zamani, N.; Mahdieh, N.; Gollob, M.H. Novel cases of pediatric sudden cardiac death secondary to TRDN mutations presenting as long QT syndrome at rest and catecholaminergic polymorphic ventricular tachycardia during exercise: The TRDN arrhythmia syndrome. Am. J. Med. Genet. Part A 2021, 185, 3433–3445.
- Altmann, H.M.; Tester, D.J.; Will, M.L.; Middha, S.; Evans, J.M.; Eckloff, B.W.; Ackerman, M.J. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: Elucidation of the triadin knockout syndrome. Circulation 2015, 131, 2051–2060.
- Song, J.; Luo, Y.; Jiang, Y.; He, J. Advances in the Molecular Genetics of Catecholaminergic Polymorphic Ventricular Tachycardia. Front. Pharmacol. 2021, 12, 2148.
- Gakenheimer-Smith, L.; Meyers, L.; Lundahl, D.; Menon, S.C.; Bunch, T.J.; Sawyer, B.L.; Tristani-Firouzi, M.; Etheridge, S.P. Expanding the phenotype of CACNA1C mutation disorders. Mol. Genet. Genom. Med. 2021, 9, e1673.
- Endres, D.; Decher, N.; Röhr, I.; Vowinkel, K.; Domschke, K.; Komlosi, K.; Tzschach, A.; Gläser, B.; Schiele, M.A.; Runge, K.; et al. New CaV1.2 channelopathy with high-functioning autism, affective disorder, severe dental enamel defects, a short QT interval, and a novel cacna1c loss-of-function mutation. Int. J. Mol. Sci. 2020, 21, 8611.
- Di Mauro, V.; Ceriotti, P.; Lodola, F.; Salvarani, N.; Modica, J.; Bang, M.L.; Mazzanti, A.; Napolitano, C.; Priori, S.G.; Catalucci, D. Peptide-Based Targeting of the L-Type Calcium Channel Corrects the Loss-of-Function Phenotype of Two Novel Mutations of the CACNA1 Gene Associated With Brugada Syndrome. Front. Physiol. 2021, 11, 1741.
- Liu, X.; Shen, Y.; Xie, J.; Bao, H.; Cao, Q.; Wan, R.; Xu, X.; Zhou, H.; Huang, L.; Xu, Z.; et al. A mutation in the CACNA1C gene leads to early repolarization syndrome with incomplete penetrance: A Chinese family study. PLoS ONE 2017, 12, e0177532.
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31.
- Mazzanti, A.; Guz, D.; Trancuccio, A.; Pagan, E.; Kukavica, D.; Chargeishvili, T.; Olivetti, N.; Biernacka, E.K.; Sacilotto, L.; Sarquella-Brugada, G.; et al. Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J. Am. Coll. Cardiol. 2020, 75, 1772–1784.
- Kalscheur, M.M.; Vaidyanathan, R.; Orland, K.M.; Abozeid, S.; Fabry, N.; Maginot, K.R.; January, C.T.; Makielski, J.C.; Eckhardt, L.L. KCNJ2 mutation causes an adrenergic-dependent rectification abnormality with calcium sensitivity and ventricular arrhythmia. Heart Rhythm 2014, 11, 885–894.
- Kimura, H.; Zhou, J.; Kawamura, M.; Itoh, H.; Mizusawa, Y.; Ding, W.G.; Wu, J.; Ohno, S.; Makiyama, T.; Miyamoto, A.; et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ. Cardiovasc. Genet. 2012, 5, 344–353.
- Rezazadeh, S.; Guo, J.; Duff, H.J.; Ferrier, R.A.; Gerull, B. Reversible Dilated Cardiomyopathy Caused by a High Burden of Ventricular Arrhythmias in Andersen-Tawil Syndrome. Can. J. Cardiol. 2016, 32, 1576.e15–1576.e18.
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; Di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807.
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142.
- Mohler, P.J.; Rivolta, I.; Napolitano, C.; LeMaillet, G.; Lambert, S.; Priori, S.G.; Bennett, V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 17533–17538.
- Giudicessi, J.R.; Ackerman, M.J. Established Loss-of-Function Variants in ANK2 -Encoded Ankyrin-B Rarely Cause a concerning Cardiac Phenotype in Humans. Circ. Genom. Precis. Med. 2020, 13, 80–82.
- Yang, R.; Walder-Christensen, K.K.; Kim, N.; Wu, D.; Lorenzo, D.N.; Badea, A.; Jiang, Y.H.; Yin, H.H.; Wetsel, W.C.; Bennett, V. ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci. USA 2019, 116, 15262–15271.
- Yamada, N.; Asano, Y.; Fujita, M.; Yamazaki, S.; Inanobe, A.; Matsuura, N.; Kobayashi, H.; Ohno, S.; Ebana, Y.; Tsukamoto, O.; et al. Mutant KCNJ3 and KCNJ5 Potassium Channels as Novel Molecular Targets in Bradyarrhythmias and Atrial Fibrillation. Circulation 2019, 139, 2157–2169.
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694.
- D’Imperio, S.; Monasky, M.M.; Micaglio, E.; Ciconte, G.; Anastasia, L.; Pappone, C. Brugada Syndrome: Warning of a Systemic Condition? Front. Cardiovasc. Med. 2021, 8, 1386.
- Boukens, B.J.; Potse, M.; Coronel, R. Fibrosis and conduction abnormalities as basis for overlap of brugada syndrome and early repolarization syndrome. Int. J. Mol. Sci. 2021, 22, 1570.
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756.
- Erdoğan, O. Coexistence of Wolff-Parkinson-White and Brugada ECG. Turk Kardiyol. Dern. Ars. 2018, 46, 433–434.
- Hothi, S.S.; Ara, F.; Timperley, J. p.Y1449C SCN5A mutation associated with overlap disorder comprising conduction disease, Brugada syndrome, and atrial flutter. J. Cardiovasc. Electrophysiol. 2015, 26, 93–97.
- Vlachos, K.; Mascia, G.; Martin, C.A.; Bazoukis, G.; Frontera, A.; Cheniti, G.; Letsas, K.P.; Efremidis, M.; Georgopoulos, S.; Gkalapis, C.; et al. Atrial fibrillation in Brugada syndrome: Current perspectives. J. Cardiovasc. Electrophysiol. 2020, 31, 975–984.
- Kewcharoen, J.; Rattanawong, P.; Kanitsoraphan, C.; Mekritthikrai, R.; Prasitlumkum, N.; Putthapiban, P.; Mekraksakit, P.; Pattison, R.J.; Vutthikraivit, W. Atrial fibrillation and risk of major arrhythmic events in Brugada syndrome: A meta-analysis. Ann. Noninvasive Electrocardiol. 2019, 24, e12676.
- Abdelghani, M.; Chapra, A.; Asaad, N.; Hayat, S. Epilepsy and Brugada Syndrome: Association or Uncommon Presentation? Heart Views 2020, 21, 114.
- Parisi, P.; Oliva, A.; Coll Vidal, M.; Partemi, S.; Campuzano, O.; Iglesias, A.; Pisani, D.; Pascali, V.L.; Paolino, M.C.; Villa, M.P.; et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013, 105, 415–418.
- Sandorfi, G.; Clemens, B.; Csanadi, Z. Electrical storm in the brain and in the heart: Epilepsy and Brugada syndrome. Mayo Clin. Proc. 2013, 88, 1167–1173.
- Camacho Velásquez, J.L.; Rivero Sanz, E.; Velazquez Benito, A.; Mauri Llerda, J.A. Epilepsia y síndrome de Brugada. Neurologia 2017, 32, 58–60.
- Sasaki, T.; Ikeda, K.; Nakajima, T.; Kawabata-Iwakawa, R.; Iizuka, T.; Dharmawan, T.; Tamura, S.; Niwamae, N.; Tange, S.; Nishiyama, M.; et al. Multiple arrhythmic and cardiomyopathic phenotypes associated with an SCN5A A735E mutation. J. Electrocardiol. 2021, 65, 122–127.
- Amarouch, M.Y.; El Hilaly, J. Inherited Cardiac Arrhythmia Syndromes: Focus on Molecular Mechanisms Underlying TRPM4 Channelopathies. Cardiovasc. Ther. 2020, 2020, 1–10.
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387.
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164.
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Sorensen, C.R.; Sheppard, M.N.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322.
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated with Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, 273–284.
- Bottillo, I.; D’Angelantonio, D.; Caputo, V.; Paiardini, A.; Lipari, M.; De Bernardo, C.; Giannarelli, D.; Pizzuti, A.; Majore, S.; Castori, M.; et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene 2016, 577, 227–235.
- Taniguchi, Y.; Miyazaki, A.; Sakaguchi, H.; Hayama, Y.; Ebishima, N.; Negishi, J.; Noritake, K.; Miyamoto, Y.; Shimizu, W.; Aiba, T.; et al. Prominent QTc prolongation in a patient with a rare variant in the cardiac ryanodine receptor gene. Heart Vessels 2017, 32, 229–233.
- Hirose, S.; Murayama, T.; Tetsuo, N.; Hoshiai, M.; Kise, H.; Yoshinaga, M.; Aoki, H.; Fukuyama, M.; Wuriyanghai, Y.; Wada, Y.; et al. Loss-of-function mutations in cardiac ryanodine receptor channel cause various types of arrhythmias including long QT syndrome. Europace 2021, euab250.
- Roston, T.M.; Guo, W.; Krahn, A.D.; Wang, R.; Van Petegem, F.; Sanatani, S.; Chen, S.R.W.; Lehman, A. A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular non-compaction and atypical catecholaminergic polymorphic ventricular tachycardia. J. Electrocardiol. 2017, 50, 227–233.
- Roston, T.M.; Wei, J.; Guo, W.; Li, Y.; Zhong, X.; Wang, R.; Estillore, J.P.; Peltenburg, P.J.; Noguer, F.R.I.; Till, J.; et al. Clinical and Functional Characterization of Ryanodine Receptor 2 Variants Implicated in Calcium-Release Deficiency Syndrome. JAMA Cardiol. 2021, e214458.
- Li, Y.; Wei, J.; Guo, W.; Sun, B.; Estillore, J.P.; Wang, R.; Yoruk, A.; Roston, T.M.; Sanatani, S.; Wilde, A.A.M.; et al. Human RyR2 (Ryanodine Receptor 2) Loss-of-Function Mutations: Clinical Phenotypes and In Vitro Characterization. Circ. Arrhythm. Electrophysiol. 2021, 14, 874–885.
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384.
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408.
- Jalloul, Y.; Refaat, M.M. Novel variants in TECRL cause catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2020, 31, 1536–1538.
- Roux-buisson, N.; Cacheux, M.; Fourest-lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767.
- Imberti, J.F.; Underwood, K.; Mazzanti, A.; Priori, S.G. Clinical Challenges in Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ. 2016, 25, 777–783.
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Recent Advances in Short QT Syndrome. Front. Cardiovasc. Med. 2018, 5.
- Thorsen, K.; Dam, V.S.; Kjaer-Sorensen, K.; Pedersen, L.N.; Skeberdis, V.A.; Jurevičius, J.; Treinys, R.; Petersen, I.M.B.S.; Nielsen, M.S.; Oxvig, C.; et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun. 2017, 8.
- Roussel, J.; Labarthe, F.; Thireau, J.; Ferro, F.; Farah, C.; Roy, J.; Horiuchi, M.; Tardieu, M.; Lefort, B.; François Benoist, J.; et al. Carnitine deficiency induces a short QT syndrome. Heart Rhythm 2016, 13, 165–174.
- Gélinas, R.; Leach, E.; Horvath, G.; Laksman, Z. Molecular Autopsy Implicates Primary Carnitine Deficiency in Sudden Unexplained Death and Reversible Short QT Syndrome. Can. J. Cardiol. 2019, 35, 1256.e1–1256.e2.
- Kessi, M.; Chen, B.; Peng, J.; Yan, F.; Yang, L.; Yin, F. Calcium channelopathies and intellectual disability: A systematic review. Orphanet J. Rare Dis. 2021, 16.
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Prim. 2020, 6, 1–22.
- Mizusawa, Y. Recent advances in genetic testing and counseling for inherited arrhythmias. J. Arrhythm. 2016, 32, 389–397.
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 507–517.
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Michael Vincent, G.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95.
- Bohnen, M.S.; Peng, G.; Robey, S.H.; Terrenoire, C.; Iyer, V.; Sampson, K.J.; Kass, R.S. Molecular pathophysiology of congenital long QT syndrome. Physiol. Rev. 2017, 97, 89–134.
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013, 127, 1009–1017.
- Crotti, L.; Johnson, C.N.; Graf, E.; De Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin mutations and life-threatening cardiac arrhythmias: Insights from the International Calmodulinopathy Registry. Eur. Heart J. 2019, 40, 2964–2975.
- Clemens, D.J.; Gray, B.; Bagnall, R.D.; Tester, D.J.; Dotzler, S.M.; Giudicessi, J.R.; Matthews, E.; Semsarian, C.; Behr, E.R.; Ackerman, M.J. Triadin Knockout Syndrome Is Absent in a Multi-Center Molecular Autopsy Cohort of Sudden Infant Death Syndrome and Sudden Unexplained Death in the Young and Is Extremely Rare in the General Population. Circ. Genom. Precis. Med. 2020, 13, 52–58.
- Korkosh, V.S.; Kiselev, A.M.; Mikhaylov, E.N.; Kostareva, A.A.; Zhorov, B.S. Atomic mechanisms of Timothy syndrome-associated mutations in calcium channel Cav1.2. Front. Physiol. 2019, 10.
- Matsuda, S.; Ohnuki, Y.; Okami, M.; Ochiai, E.; Yamada, S.; Takahashi, K.; Osawa, M.; Okami, K.; Iida, M.; Mochizuki, H. Jervell and Lange-Nielsen syndrome with novel KCNQ1 and additional gene mutations. Hum. Genome Var. 2020, 7.
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 2020, 142, 324–338.
- Napolitano, C.; Mazzanti, A.; Priori, S.G. Genetic risk stratification in cardiac arrhythmias. Curr. Opin. Cardiol. 2018, 33, 298–303.
- Ronchi, C.; Bernardi, J.; Mura, M.; Stefanello, M.; Badone, B.; Rocchetti, M.; Crotti, L.; Brink, P.; Schwartz, P.J.; Gnecchi, M.; et al. NOS1AP polymorphisms reduce NOS1 activity and interact with prolonged repolarization in arrhythmogenesis. Cardiovasc. Res. 2021, 117, 472–483.
- Kääb, S.; Crawford, D.C.; Sinner, M.F.; Behr, E.R.; Kannankeril, P.J.; Wilde, A.A.M.; Bezzina, C.R.; Schulze-Bahr, E.; Guicheney, P.; Bishopric, N.H.; et al. A large candidate gene survey identifies the KCNE1D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ. Cardiovasc. Genet. 2012, 5, 91–99.
- Sesti, F.; Abbott, G.W.; Wei, J.; Murray, K.T.; Saksena, S.; Schwartz, P.J.; Priori, S.G.; Roden, D.M.; George, A.L.; Goldstein, S.A.N. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 2000, 97, 10613–10618.
- Cheng, J.; Tester, D.J.; Tan, B.H.; Valdivia, C.R.; Kroboth, S.; Ye, B.; January, C.T.; Ackerman, M.J.; Makielski, J.C. The common African American polymorphism SCN5A-S1103Y interacts with mutation SCN5A-R680h to increase late Na current. Physiol. Genom. 2011, 43, 461–466.
- Roberts, J.D.; Krahn, A.D.; Ackerman, M.J.; Rohatgi, R.K.; Moss, A.J.; Nazer, B.; Tadros, R.; Gerull, B.; Sanatani, S.; Wijeyeratne, Y.D.; et al. Loss-of-Function KCNE2 Variants: True Monogenic Culprits of Long-QT Syndrome or Proarrhythmic Variants Requiring Secondary Provocation? Circ. Arrhythm. Electrophysiol. 2017, 10.
- El-Sherif, N.; Turitto, G.; Boutjdir, M. Acquired long QT syndrome and torsade de pointes. Pacing Clin. Electrophysiol. 2018, 41, 414–421.
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europe. Europace 2015, 17, 1601–1687.
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993, 88, 782–784.
- Goldenberg, I.; Horr, S.; Moss, A.J.; Lopes, C.M.; Barsheshet, A.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J. Am. Coll. Cardiol. 2011, 57, 51–59.
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk Stratification in the Long-QT Syndrome. N. Engl. J. Med. 2003, 348, 1866–1874.
- Mazzanti, A.; Maragna, R.; Vacanti, G.; Monteforte, N.; Bloise, R.; Marino, M.; Braghieri, L.; Gambelli, P.; Memmi, M.; Pagan, E.; et al. Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients With Long QT Syndrome. J. Am. Coll. Cardiol. 2018, 71, 1663–1671.
- Vink, A.S.; Clur, S.A.B.; Geskus, R.B.; Blank, A.C.; De Kezel, C.C.A.; Yoshinaga, M.; Hofman, N.; Wilde, A.A.M.; Blom, N.A. Effect of Age and Sex on the QTc Interval in Children and Adolescents with Type 1 and 2 Long-QT Syndrome. Circ. Arrhythm. Electrophysiol. 2017, 10.
- Jons, C.; Moss, A.J.; Goldenberg, I.; Liu, J.; McNitt, S.; Zareba, W.; Qi, M.; Robinson, J.L. Risk of Fatal Arrhythmic Events in Long QT Syndrome Patients After Syncope. J. Am. Coll. Cardiol. 2010, 55, 783–788.
- Sharma, N.; Cortez, D.; Disori, K.; Imundo, J.R.; Beck, M. A Review of Long QT Syndrome: Everything a Hospitalist Should Know. Hosp. Pediatr. 2020, 10, 369–375.
- Moore, J.P.; Gallotti, R.G.; Shannon, K.M.; Bos, J.M.; Sadeghi, E.; Strasburger, J.F.; Wakai, R.T.; Horigome, H.; Clur, S.A.; Hill, A.C.; et al. Genotype Predicts Outcomes in Fetuses and Neonates With Severe Congenital Long QT Syndrome. JACC Clin. Electrophysiol. 2020, 6, 1561–1570.
- Seth, R.; Moss, A.J.; McNitt, S.; Zareba, W.; Andrews, M.L.; Qi, M.; Robinson, J.L.; Goldenberg, I.; Ackerman, M.J.; Benhorin, J.; et al. Long QT Syndrome and Pregnancy. J. Am. Coll. Cardiol. 2007, 49, 1092–1098.
- Shimizu, W.; Makimoto, H.; Yamagata, K.; Kamakura, T.; Wada, M.; Miyamoto, K.; Inoue-Yamada, Y.; Okamura, H.; Ishibashi, K.; Noda, T.; et al. Association of Genetic and Clinical Aspects of Congenital Long QT Syndrome with Life-Threatening Arrhythmias in Japanese Patients. JAMA Cardiol. 2019, 4, 246–254.
- Amin, A.S.; Herfst, L.J.; Delisle, B.P.; Klemens, C.A.; Rook, M.B.; Bezzina, C.R.; Underkofler, H.A.S.; Holzem, K.M.; Ruijter, J.M.; Tan, H.L.; et al. Fever-induced QTc prolongation and ventricular arrhythmias in individuals with type 2 congenital long QT syndrome. J. Clin. Investig. 2008, 118, 2552–2561.
- Giudicessi, J.R.; Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 2013, 161, 1–14.
- Crotti, L.; Spazzolini, C.; Schwartz, P.J.; Shimizu, W.; Denjoy, I.; Schulze-Bahr, E.; Zaklyazminskaya, E.V.; Swan, H.; Ackerman, M.J.; Moss, A.J.; et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: Toward a mutation-specific risk stratification. Circulation 2007, 116, 2366–2375.
- Priest, J.R.; Gawad, C.; Kahlig, K.M.; Yuf, J.K.; O’Hara, T.; Boyle, P.M.; Rajamani, S.; Clark, M.J.; Garcia, S.T.K.; Ceresnak, S.; et al. Early somatic mosaicism is a rare cause of long-QT syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 11555–11560.
- Napolitano, C.; Novelli, V.; Francis, M.D.; Priori, S.G. Genetic modulators of the phenotype in the long QT syndrome: State of the art and clinical impact. Curr. Opin. Genet. Dev. 2015, 33, 17–24.
- Riuró, H.; Campuzano, O.; Berne, P.; Arbelo, E.; Iglesias, A.; Pérez-Serra, A.; Coll-Vidal, M.; Partemi, S.; Mademont-Soler, I.; Picó, F.; et al. Genetic analysis, in silico prediction, and family segregation in long QT syndrome. Eur. J. Hum. Genet. 2015, 23, 79–85.
- Marín, P.N.; Jiménez-Jáimez, J.; Tinaquero, D.; Alfayate, S.; Utrilla, R.G.; Del Rey, M.D.M.R.V.; Perin, F.; Sarquella-Brugada, G.; Monserrat, L.; Brugada, J.; et al. Digenic Heterozigosity in SCN5A and CACNA1C Explains the Variable Expressivity of the Long QT Phenotype in a Spanish Family. Rev. Española Cardiol. (Engl. Ed.) 2019, 72, 324–332.
- Eddy, C.A.; MacCormick, J.M.; Chung, S.K.; Crawford, J.R.; Love, D.R.; Rees, M.I.; Skinner, J.R.; Shelling, A.N. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm 2008, 5, 1275–1281.
- Barc, J.; Briec, F.; Schmitt, S.; Kyndt, F.; Le Cunff, M.; Baron, E.; Vieyres, C.; Sacher, F.; Redon, R.; Le Caignec, C.; et al. Screening for copy number variation in genes associated with the long QT syndrome: Clinical relevance. J. Am. Coll. Cardiol. 2011, 57, 40–47.
- Stattin, E.L.; Boström, I.M.; Winbo, A.; Cederquist, K.; Jonasson, J.; Jonsson, B.A.; Diamant, U.B.; Jensen, S.M.; Rydberg, A.; Norberg, A. Founder mutations characterise the mutation panorama in 200 Swedish index cases referred for Long QT syndrome genetic testing. BMC Cardiovasc. Disord. 2012, 12, 95.
- Campuzano, O.; Sarquella-Brugada, G.; Mademont-Soler, I.; Allegue, C.; Cesar, S.; Ferrer-Costa, C.; Coll, M.; Mates, J.; Iglesias, A.; Brugada, J.; et al. Identification of genetic alterations, as causative genetic defects in long QT syndrome, using next generation sequencing technology. PLoS ONE 2014, 9, e114894.
- Mates, J.; Mademont-Soler, I.; Del Olmo, B.; Ferrer-Costa, C.; Coll, M.; Pérez-Serra, A.; Picó, F.; Allegue, C.; Fernandez-Falgueras, A.; Álvarez, P.; et al. Role of copy number variants in sudden cardiac death and related diseases: Genetic analysis and translation into clinical practice. Eur. J. Hum. Genet. 2018, 26, 1014–1025.
- Earle, N.; Crawford, J.; Smith, W.; Hayes, I.; Shelling, A.; Hood, M.; Stiles, M.; Maxwell, F.; Heaven, D.; Love, D.R.; et al. Community detection of long QT syndrome with a clinical registry: An alternative to ECG screening programs? Heart Rhythm 2013, 10, 233–238.
- Sarquella-Brugada, G.; Cesar, S.; Zambrano, M.D.; Fernandez-Falgueras, A.; Fiol, V.; Iglesias, A.; Torres, F.; Garcia-Algar, O.; Arbelo, E.; Brugada, J.; et al. Electrocardiographic Assessment and Genetic Analysis in Neonates: A Current Topic of Discussion. Curr. Cardiol. Rev. 2018, 15, 30–37.
- Gomez, A.T.; Prutkin, J.M.; Rao, A.L. Evaluation and Management of Athletes With Long QT Syndrome: An Evolved Paradigm. Sports Health 2016, 8, 527–535.
- Ahluwalia, N.; Raju, H. Assessment of the QT Interval in Athletes: Red Flags and Pitfalls. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 1–12.
- Wilde, A.A.M.; Moss, A.J.; Kaufman, E.S.; Shimizu, W.; Peterson, D.R.; Benhorin, J.; Lopes, C.; Towbin, J.A.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882.
- Yang, Y.; Ly, T.T.; Li, S.Y.; Zhang, P. Sodium channel blockers in the management of long QT syndrome types 3 and 2: A system review and meta-analysis. J. Cardiovasc. Electrophysiol. 2021, 32, 3057–3067.
- Ackerman, M.J.; Priori, S.G.; Dubin, A.M.; Kowey, P.; Linker, N.J.; Slotwiner, D.; Triedman, J.; Van Hare, G.F.; Gold, M.R. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: Are all beta-blockers equivalent? Heart Rhythm 2017, 14, e41–e44.
- Waddell-Smith, K.E.; Earle, N.; Skinner, J.R. Must every child with long QT syndrome take a beta blocker? Arch. Dis. Child. 2015, 100, 279–282.
- Niaz, T.; Bos, J.M.; Sorensen, K.B.; Moir, C.; Ackerman, M.J. Left Cardiac Sympathetic Denervation Monotherapy in Patients with Congenital Long QT Syndrome. Circ. Arrhythm. Electrophysiol. 2020, 13, e008830.
- Schwartz, P.J.; Spazzolini, C.; Priori, S.G.; Crotti, L.; Vicentini, A.; Landolina, M.; Gasparini, M.; Wilde, A.A.M.; Knops, R.E.; Denjoy, I.; et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them?: Data from the European Long-QT syndrome implantable cardioverter-defibrillator (LQTS ICD) registry. Circulation 2010, 122, 1272–1282.
- Shah, M.J.; Silka, M.J.; Silva, J.N.A.; Balaji, S.; Beach, C.M.; Benjamin, M.N.; Berul, C.I.; Cannon, B.; Cecchin, F.; Cohen, M.I.; et al. 2021 PACES Expert Consensus Statement on the Indications and Management of Cardiovascular Implantable Electronic Devices in Pediatric Patients. Heart Rhythm 2021, 18, 1888–1924.
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059.
- Mizusawa, Y.; Wilde, A.A.M. Brugada syndrome. Circ. Arrhythm. Electrophysiol. 2012, 5, 606–616.
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender Differences in Clinical Manifestations of Brugada Syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573.
- Milman, A.; Andorin, A.; Gourraud, J.B.; Sacher, F.; Mabo, P.; Kim, S.H.; Maeda, S.; Takahashi, Y.; Kamakura, T.; Aiba, T.; et al. Age of First Arrhythmic Event in Brugada Syndrome: Data from the SABRUS (Survey on Arrhythmic Events in Brugada Syndrome) in 678 Patients. Circ. Arrhythm. Electrophysiol. 2017, 10, e005222.
- Raharjo, S.B.; Maulana, R.; Maghfirah, I.; Alzahra, F.; Putrinarita, A.D.; Hanafy, D.A.; Yuniadi, Y. SCN5A gene mutations and the risk of ventricular fibrillation and syncope in brugada syndrome patients: A meta-analysis. J. Arrhythm. 2018, 34, 473–477.
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396.
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada syndrome: Oligogenic or mendelian disease? Int. J. Mol. Sci. 2020, 21, 1687.
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Update on genetic basis of Brugada syndrome: Monogenic, polygenic or oligogenic? Int. J. Mol. Sci. 2020, 21, 7155.
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C. Genetic testing for inherited cardiovascular diseases: A scientific statement from the american heart association. Circ. Genom. Precis. Med. 2020, 13, 373–385.
- Crotti, L.; Marcou, C.A.; Tester, D.J.; Castelletti, S.; Giudicessi, J.R.; Torchio, M.; Medeiros-Domingo, A.; Simone, S.; Will, M.L.; Dagradi, F.; et al. Spectrum and prevalence of mutations involving BrS1-Through BrS12-susceptibility genes in a cohort of unrelated patients referred for brugada syndrome genetic testing: Implications for genetic testing. J. Am. Coll. Cardiol. 2012, 60, 1410–1418.
- Abbott, G.W. KCNE4 and KCNE5: K(+) channel regulation and cardiac arrhythmogenesis. Gene 2016, 593, 249–260.
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109.
- Michowitz, Y.; Milman, A.; Sarquella-Brugada, G.; Andorin, A.; Champagne, J.; Postema, P.G.; Casado-Arroyo, R.; Leshem, E.; Juang, J.J.; Giustetto, C.; et al. Fever-related arrhythmic events in the multicenter Survey on Arrhythmic Events in Brugada Syndrome. Heart Rhythm 2018, 15, 1394–1401.
- Chen, X.; Zhao, H.; Sun, L.; Zhu, W.; Zhang, F. Electrocardiogram Characteristics and Arrhythmic Events during Fever in Patients with Fever-Induced Brugada Syndrome. Cardiology 2020, 145, 130–135.
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.G.; Verkerk, A.O.; De Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777.
- Nademanee, K.; Raju, H.; De Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986.
- Pieroni, M.; Notarstefano, P.; Oliva, A.; Campuzano, O.; Santangeli, P.; Coll, M.; Nesti, M.; Carnevali, A.; Fraticelli, A.; Iglesias, A.; et al. Electroanatomic and Pathologic Right Ventricular Outflow Tract Abnormalities in Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 72, 2747–2757.
- Michowitz, Y.; Milman, A.; Andorin, A.; Sarquella-Brugada, G.; Gonzalez Corcia, M.C.; Gourraud, J.B.; Conte, G.; Sacher, F.; Juang, J.J.M.; Kim, S.H.; et al. Characterization and Management of Arrhythmic Events in Young Patients With Brugada Syndrome. J. Am. Coll. Cardiol. 2019, 73, 1756–1765.
- Gourraud, J.-B.; Barc, J.; Thollet, A.; Le Marec, H.; Probst, V. Brugada syndrome: Diagnosis, risk stratification and management. Arch. Cardiovasc. Dis. 2017, 110, 188–195.
- Gonzalez Corcia, M.C.; Sieira, J.; Sarkozy, A.; De Asmundis, C.; Chierchia, G.B.; Hernandez Ojeda, J.; Pappaert, G.; Brugada, P. Brugada syndrome in the young: An assessment of risk factors predicting future events. Europace 2017, 19, 1864–1873.
- Milman, A.; Gourraud, J.B.; Andorin, A.; Postema, P.G.; Sacher, F.; Mabo, P.; Conte, G.; Giustetto, C.; Sarquella-Brugada, G.; Hochstadt, A.; et al. Gender differences in patients with Brugada syndrome and arrhythmic events: Data from a survey on arrhythmic events in 678 patients. Heart Rhythm 2018, 15, 1457–1465.
- Priori, S.G.; Gasparini, M.; Napolitano, C.; Della Bella, P.; Ottonelli, A.G.; Sassone, B.; Giordano, U.; Pappone, C.; Mascioli, G.; Rossetti, G.; et al. Risk Stratification in Brugada Syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) Registry. J. Am. Coll. Cardiol. 2012, 59, 37–45.
- Probst, V.; Veltmann, C.; Eckardt, L.; Meregalli, P.G.; Gaita, F.; Tan, H.L.; Babuty, D.; Sacher, F.; Giustetto, C.; Schulze-Bahr, E.; et al. Long-term prognosis of patients diagnosed with brugada syndrome: Results from the finger brugada syndrome registry. Circulation 2010, 121, 635–643.
- Morita, H.; Kusano, K.F.; Miura, D.; Nagase, S.; Nakamura, K.; Morita, S.T.; Ohe, T.; Zipes, D.P.; Wu, J. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation 2008, 118, 1697–1704.
- Argenziano, M.; Antzelevitch, C. Recent advances in the treatment of Brugada syndrome. Expert Rev. Cardiovasc. Ther. 2018, 16, 387–404.
- Jongman, J.K.; Jepkes-Bruin, N.; Ramdat Misier, A.R.; Beukema, W.P.; Delnoy, P.P.H.M.; Oude Luttikhuis, H.; Dambrink, J.H.E.; Hoorntje, J.C.A.; Elvan, A. Electrical storms in Brugada syndrome successfully treated with isoproterenol infusion and quinidine orally. Neth. Heart J. 2007, 15, 151–154.
- Gonzalez Corcia, M.C.; Sieira, J.; Pappaert, G.; de Asmundis, C.; Chierchia, G.B.; La Meir, M.; Sarkozy, A.; Brugada, P. Implantable Cardioverter-Defibrillators in Children and Adolescents With Brugada Syndrome. J. Am. Coll. Cardiol. 2018, 71, 148–157.
- Hernandez-Ojeda, J.; Arbelo, E.; Borras, R.; Berne, P.; Tolosana, J.M.; Gomez-Juanatey, A.; Berruezo, A.; Campuzano, O.; Sarquella-Brugada, G.; Mont, L.; et al. Patients With Brugada Syndrome and Implanted Cardioverter-Defibrillators: Long-Term Follow-Up. J. Am. Coll. Cardiol. 2017, 70, 1991–2002.
- Rizzo, A.; de Asmundis, C.; Brugada, P.; La Meir, M.; Chierchia, G.B. Ablation for the treatment of Brugada syndrome: Current status and future prospects. Expert Rev. Med. Devices 2020, 17, 123–130.
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000, 102, 2509–2515.
- Probst, V.; Wilde, A.A.M.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A Mutations and the role of genetic background in the pathophysiology of brugada syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557.
- Mates, J.; Mademont-Soler, I.; Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; García-Álvarez, A.; Jordà, P.; Toro, R.; Coll, M.; et al. Sudden Cardiac Death and Copy Number Variants: What Do We Know after 10 Years of Genetic Analysis? Forensic Sci. Int. Genet. 2020, 47, 102281.
- Gonzalez Corcia, M.C.; Brugada, P. Family Screening for Brugada Syndrome in Asymptomatic Young Patients. Is it Better not to Know? Pediatr. Cardiol. 2017, 38, 1313–1314.
- Therasse, D.; Sacher, F.; Petit, B.; Babuty, D.; Mabo, P.; Martins, R.; Jesel, L.; Maury, P.; Pasquie, J.L.; Mansourati, J.; et al. Sodium-channel blocker challenge in the familial screening of Brugada syndrome: Safety and predictors of positivity. Heart Rhythm 2017, 14, 1442–1448.
- Rizzo, A.; Borio, G.; Sieira, J.; Van Dooren, S.; Overeinder, I.; Bala, G.; Pappaert, G.; Maj, R.; Osório, T.G.; Terasawa, M.; et al. Ajmaline Testing and the Brugada Syndrome. Am. J. Cardiol. 2020, 135, 91–98.
- Guerrier, K.; Kwiatkowski, D.; Czosek, R.J.; Spar, D.S.; Anderson, J.B.; Knilans, T.K. Short QT Interval Prevalence and Clinical Outcomes in a Pediatric Population. Circ. Arrhythm. Electrophysiol. 2015, 8, 1460–1464.
- Bjerregaard, P. Diagnosis and management of short QT syndrome. Heart Rhythm 2018, 15, 1261–1267.
- El-Battrawy, I.; Besler, J.; Liebe, V.; Schimpf, R.; Tülümen, E.; Rudic, B.; Lang, S.; Wolpert, C.; Zhou, X.; Akin, I.; et al. Long-term follow-up of patients with short QT syndrome: Clinical profile and outcome. J. Am. Heart Assoc. 2018, 7, e010073.
- Mazzanti, A.; Kanthan, A.; Monteforte, N.; Memmi, M.; Bloise, R.; Novelli, V.; Miceli, C.; O’Rourke, S.; Borio, G.; Zienciuk-Krajka, A.; et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 2014, 63, 1300–1308.
- Hu, D.; Li, Y.; Zhang, J.; Pfeiffer, R.; Gollob, M.H.; Healey, J.; Harrell, D.T.; Makita, N.; Abe, H.; Sun, Y.; et al. The Phenotypic Spectrum of a Mutation Hotspot Responsible for the Short QT Syndrome. JACC Clin. Electrophysiol. 2017, 3, 727–743.
- Campuzano, O.; Fernandez-Falgueras, A.; Lemus, X.; Sarquella-Brugada, G.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Jordà, P.; Perez-Serra, A.; et al. Short QT syndrome: A comprehensive genetic interpretation and clinical translation of rare variants. J. Clin. Med. 2019, 8, 1035.
- Harrell, D.T.; Ashihara, T.; Ishikawa, T.; Tominaga, I.; Mazzanti, A.; Takahashi, K.; Oginosawa, Y.; Abe, H.; Maemura, K.; Sumitomo, N.; et al. Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. Int. J. Cardiol. 2015, 190, 393–402.
- Dewi, I.P.; Dharmadjati, B.B. Short QT syndrome: The current evidences of diagnosis and management. J. Arrhythm. 2020, 36, 962–966.
- Chinese Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Available online: https://pubmed.ncbi.nlm.nih.gov/25876716/ (accessed on 1 November 2021).
- Hong, K.; Piper, D.R.; Diaz-Valdecantos, A.; Brugada, J.; Oliva, A.; Burashnikov, E.; Santos-De-Soto, J.; Grueso-Montero, J.; Diaz-Enfante, E.; Brugada, P.; et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 2005, 68, 433–440.
- Sarquella-Brugada, G.; Campuzano, O.; Iglesias, A.; Grueso, J.; Bradley, D.J.; Kerst, G.; Shmorhun, D.; Brugada, J.; Brugada, R. Short QT and atrial fibrillation: A KCNQ1 mutation-specific disease. Late follow-up in three unrelated children. HeartRhythm Case Rep. 2015, 1, 193–197.
- Providência, R.; Karim, N.; Srinivasan, N.; Honarbakhsh, S.; Ferreira, M.J.V.; Gonçalves, L.; Marijon, E.; Lambiase, P.D. Impact of QTc formulae in the prevalence of short corrected QT interval and impact on probability and diagnosis of short QT syndrome. Heart 2018, 104, 502–508.
- Hazeki, D.; Ninomiya, Y.; Ueno, K.; Yoshinaga, M. Tentative screening criteria for short QT interval in children and adolescents. Circ. J. 2018, 82, 2627–2633.
- Fan, X.; Yang, G.; Kowitx, J.; Duru, F.; Saguner, A.M.; Akin, I.; Zhou, X.; El-Battrawy, I. Preclinical short QT syndrome models: Studying the phenotype and drug-screening. Europace 2021, euab214.
- Suzuki, H.; Horie, M.; Ozawa, J.; Sumitomo, N.; Ohno, S.; Hoshino, K.; Ehara, E.; Takahashi, K.; Maeda, Y.; Yoshinaga, M.; et al. Novel electrocardiographic criteria for short QT syndrome in children and adolescents. Europace 2021, 23, 2029–2038.
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963.
- Roston, T.M.; Yuchi, Z.; Kannankeril, P.J.; Hathaway, J.; Vinocur, J.M.; Etheridge, S.P.; Potts, J.E.; Maginot, K.R.; Salerno, J.C.; Cohen, M.I.; et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018, 20, 541–547.
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74.
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.M.; Klug, D.; Hayashi, M.; Takatsuki, S.; Villain, E.; et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009, 119, 2426–2434.
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutataions in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200.
- Pérez-Riera, A.R.; Barbosa-Barros, R.; de Rezende Barbosa, M.P.C.; Daminello-Raimundo, R.; de Lucca, A.A.; de Abreu, L.C. Catecholaminergic polymorphic ventricular tachycardia, an update. Ann. Noninvasive Electrocardiol. 2018, 23, e12512.
- Gaburjakova, M.; Bal, N.C.; Gaburjakova, J.; Periasamy, M. Functional interaction between calsequestrin and ryanodine receptor in the heart. Cell. Mol. Life Sci. 2013, 70, 2935–2945.
- Webster, G.; Aburawi, E.H.; Chaix, M.A.; Chandler, S.; Foo, R.; Islam, A.K.M.M.; A E Kammeraad, J.; Rioux, J.D.; Al-Gazali, L.; Sayeed, Z.; et al. Life-threatening arrhythmias with autosomal recessive TECRL variants. Europace 2021, 23, 781–788.
- Nyegaard, M.; Overgaard, M.T.; Sondergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712.
- Gomez-Hurtado, N.; Boczek, N.J.; Kryshtal, D.O.; Johnson, C.N.; Sun, J.; Nitu, F.R.; Cornea, R.L.; Chazin, W.J.; Calvert, M.L.; Tester, D.J.; et al. Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ. Arrhythm. Electrophysiol. 2016, 9, 9.
- Broendberg, A.K.; Nielsen, J.C.; Bjerre, J.; Pedersen, L.N.; Kristensen, J.; Henriksen, F.L.; Bundgaard, H.; Jensen, H.K. Nationwide experience of catecholaminergic polymorphic ventricular tachycardia caused by RyR2 mutations. Heart 2017, 103, 901–909.
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490.
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Exome analysis in 34 sudden unexplained death (SUD) victims mainly identified variants in channelopathy-associated genes. Int. J. Legal Med. 2018, 132, 1057–1065.
- Van Der Werf, C.; Nederend, I.; Hofman, N.; Van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.E.; Alings, A.M.W.; Bosker, H.A.; Bracke, F.A.; Van Dan Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756.
- Kallas, D.; Roston, T.M.; Franciosi, S.; Brett, L.; Lieve, K.V.; Kwok, S.-Y.; Kannankeril, P.J.; Krahn, A.D.; LaPage, M.J.; Etheridge, S.; et al. Evaluation of age at symptom onset, proband status, and sex as predictors of disease severity in pediatric catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2021, 18, 1825–1832.
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic Polymorphic Ventricular Tachycardia in Children: Analysis of Therapeutic Strategies and Outcomes from an International Multicenter Registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642.
- McNamara, C.; Cullen, P.; Rackauskas, M.; Kelly, R.; O’Sullivan, K.E.; Galvin, J.; Eaton, D. Left cardiac sympathetic denervation: Case series and technical report. Ir. J. Med. Sci. 2017, 186, 607–613.
- Roston, T.M.; Jones, K.; Hawkins, N.M.; Bos, J.M.; Schwartz, P.J.; Perry, F.; Ackerman, M.J.; Laksman, Z.W.; Kaul, P.; Lieve, K.V.; et al. Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: A systematic review. Heart Rhythm 2018, 15, 1791–1799.
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.A.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870.
- Campbell, M.; Czosek, R.J.; Hinton, R.B.; Miller, E.M. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am. J. Med. Genet. Part A 2015, 167, 2197–2200.
- Bhuiyan, Z.A.; Van Den Berg, M.P.; Van Tintelen, J.P.; Bink-Boelkens, M.T.E.; Wiesfeld, A.C.P.; Alders, M.; Postma, A.V.; Van Langen, I.; Mannens, M.M.A.M.; Wilde, A.A.M. Expanding spectrum of human RYR2-related disease: New electrocardiographic, structural, and genetic features. Circulation 2007, 116, 1569–1576.
- Persampieri, S.; Pilato, C.A.; Sommariva, E.; Maione, A.S.; Stadiotti, I.; Ranalletta, A.; Torchio, M.; Dello Russo, A.; Basso, C.; Pompilio, G.; et al. Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation. Genes (Basel) 2020, 11, 571.
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Béziau, D.M.; Marsman, R.F.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal Ectopic Purkinje-Related Premature Contractions: A New SCN5A-Related Cardiac Channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156.
- Amin, A.S. SCN5a overlap syndromes-This episode: Long QT syndrome type 3 meets multifocal ectopic Purkinje-related premature contractions. Heart Rhythm 2020, 17, 1777–1778.
- Swan, H.; Amarouch, M.Y.; Leinonen, J.; Marjamaa, A.; Kucera, J.P.; Laitinen-Forsblom, P.J.; Lahtinen, A.M.; Palotie, A.; Kontula, K.; Toivonen, L.; et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ. Cardiovasc. Genet. 2014, 7, 771–781.
- Ghovanloo, M.R.; Atallah, J.; Escudero, C.A.; Ruben, P.C. Biophysical Characterization of a Novel SCN5A Mutation Associated with an Atypical Phenotype of Atrial and Ventricular Arrhythmias and Sudden Death. Front. Physiol. 2020, 11, 11.
- Wilde, A.A.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC. Clin. Electrophysiol. 2018, 4, 569–579.
- Hayashi, H.; Sumiyoshi, M.; Nakazato, Y.; Daida, H. Brugada syndrome and sinus node dysfunction. J. Arrhythm. 2018, 34, 216–221.
- Shan, L.; Makita, N.; Xing, Y.; Watanabe, S.; Futatani, T.; Ye, F.; Saito, K.; Ibuki, K.; Watanabe, K.; Hirono, K.; et al. SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol. Genet. Metab. 2008, 93, 468–474.
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Cáceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J. Am. Heart Assoc. 2016, 5, 5.
- Sandhu, A.; Borne, R.T.; Mam, C.; Bunch, T.J.; Aleong, R.G. Double jeopardy: Long QT3 and Brugada syndromes. Clin. Case Rep. 2017, 5, 1315–1319.
- Bekke, R.M.; Isaacs, A.; Barysenka, A.; Hoos, M.B.; Jongbloed, J.D.; Hoorntje, J.C.; Patelski, A.S.; Enden, A.T.H.-V.D.; Wijngaard, A.V.D.; Stoll, M.; et al. Heritability in a SCN5A-mutation founder population with increased female susceptibility to non-nocturnal ventricular tachyarrhythmia and sudden cardiac death. Heart Rhythm 2017, 14, 1873–1881.
- Bezzina, C.; Veldkamp, M.W.; Van Den Berg, M.P.; Postma, A.V.; Rook, M.B.; Viersma, J.W.; Van Langen, I.M.; Tan-Sindhunata, G.; Bink-Boelkens, M.T.E.; Van Der Hout, A.H.; et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 1999, 85, 1206–1213.
- Coll, M.; Striano, P.; Ferrer-Costa, C.; Campuzano, O.; Matés, J.; Del Olmo, B.; Iglesias, A.; Pérez-Serra, A.; Mademont, I.; Picó, F.; et al. Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PLoS ONE 2017, 12, e0189618.
- Coll, M.; Oliva, A.; Grassi, S.; Brugada, R.; Campuzano, O. Update on the Genetic Basis of Sudden Unexpected Death in Epilepsy. Int. J. Mol. Sci. 2019, 20, 1979.
- Chahal, C.A.A.; Salloum, M.N.; Alahdab, F.; Gottwald, J.A.; Tester, D.J.; Anwer, L.A.; So, E.L.; Murad, M.H.; Louis, E.K.S.; Ackerman, M.J.; et al. Systematic Review of the Genetics of Sudden Unexpected Death in Epilepsy: Potential Overlap With Sudden Cardiac Death and Arrhythmia-Related Genes. J. Am. Heart Assoc. 2020, 9, e012264.
- Kim, S.H.; Nam, G.B.; Yun, S.C.; Choi, H.O.; Choi, K.J.; Joung, B.; Pak, H.N.; Lee, M.H.; Kim, S.S.; Park, S.J.; et al. Variants of Brugada Syndrome and Early Repolarization Syndrome: An Expanded Concept of J-Wave Syndrome. Pacing Clin. Electrophysiol. 2017, 40, 162–174.
- Scheirlynck, E.; Chivulescu, M.; Lie, Ø.H.; Scheirlynck, E.; Chivulescu, M.; Øyvind, L.; Motoc, A.; Koulalis, J.; de Asmundis, C.; Sieira, J.; et al. Worse Prognosis in Brugada Syndrome Patients with Arrhythmogenic Cardiomyopathy Features. JACC Clin. Electrophysiol. 2020, 6, 1353–1363.
- Rabah, H.; Rabah, A. Arrhythmogenic Right Ventricular Dysplasia and Brugada Syndrome Overlap. Cureus 2021, 13, 13.
- Peters, S.; Trümmel, M.; Denecke, S.; Koehler, B. Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 2004, 95, 207–210.
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005, 112, 3680–3687.
- Hoogendijk, M.G. Diagnostic Dilemmas: Overlapping Features of Brugada Syndrome and Arrhythmogenic Right Ventricular Cardiomyopathy. Front. Physiol. 2012, 3, 144.
- Priori, S.G.; Napolitano, C.; Schwartz, P.J.; Bloise, R.; Crotti, L.; Ronchetti, E. The elusive link between LQT3 and brugada syndrome: The role of flecainide challenge. Circulation 2000, 102, 945–947.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
595
Revisions:
2 times
(View History)
Update Date:
12 Jan 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No