 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peter A. Keyel | + 1896 word(s) | 1896 | 2020-08-25 10:47:00 | | | |
| 2 | Felix Wu | -23 word(s) | 1873 | 2020-11-09 02:45:12 | | | | |
| 3 | Felix Wu | -23 word(s) | 1873 | 2020-11-09 02:49:49 | | |
Video Upload Options
Cholesterol-dependent cytolysins (CDCs) are key virulence factors involved in many lethal bacterial infections, including pneumonia, necrotizing soft tissue infections, bacterial meningitis, and miscarriage. Host responses to these diseases involve myeloid cells, especially macrophages. Macrophages use several systems to detect and respond to cholesterol-dependent cytolysins, including membrane repair, mitogen-activated protein (MAP) kinase signaling, phagocytosis, cytokine production, and activation of the adaptive immune system. However, CDCs also promote immune evasion by silencing and/or destroying myeloid cells. While there are many common themes between the various CDCs, each CDC also possesses specific features to optimally benefit the pathogen producing it.
1. Introduction
Cholesterol-dependent cytolysins (CDCs) are a subset of pore-forming toxins that serve as key virulence factors for a wide range of lethal and opportunistic Gram-positive bacterial pathogens that collectively infect or invade nearly all parts of the human body. Consequently, hosts attempt to eliminate these pathogens with both general and tissue-specific approaches. One common approach that has tissue-specific flexibility is activation and polarization of macrophages. Macrophages coordinate the local tissue response with cytokines and can directly eliminate bacteria through phagocytosis and secretion of reactive oxygen/nitrogen species. They further promote wound repair and restore the tissue to homeostasis. As a result, pathogenic bacteria target macrophages for elimination, reprogramming, or shelter. CDCs figure prominently in many of these attempts. This review explores both general and specific molecular mechanisms used by CDCs to kill, control, or evade macrophages.
2. CDC Interactions with Macrophages
2.1. Cytokine Production in Response to CDCs
One key consequence of activating the signaling pathways, especially the p38 pathway, is production of pro-inflammatory cytokines. The major pro-inflammatory cytokines induced by CDC challenge are Tumor Necrosis Factor α (TNFα,) Interleukin (IL)-1β, IL-6, and IL-8, though other cytokines and chemokines are also CDC-dependent. However, one challenge with CDC-dependent cytokine production is ascertaining if the cytokine is produced by signaling pathways directly stimulated by the CDC or indirectly by other danger- or pathogen-associated molecular patterns (DAMPs and PAMPs). Many studies attributing cytokine production to CDCs used bacteria with and without deletion of the CDC. Since CDCs are often key to bacterial virulence, it is difficult to discern if changes in pro-inflammatory cytokine production are due to overall reduced virulence or directly due to the toxin. Pure CDCs may contain contaminants, such as toll-like receptor (TLR) ligands, which complicate interpretation of data. Often studies do not include inactive toxins to control for the presence of any contaminants in purified toxins. Cellular damage by CDCs further releases several DAMPs, including IL-1α, ATP, and high mobility group box 1 protein (HMGB1) [1][2][3][4], which may exert autocrine and paracrine effects on cells, including TLR engagement and pro-inflammatory cytokine production. These effects may be especially apparent when longer (12+ h) time points are used. Finally, signaling pathways can be activated independently of plasma membrane receptors by the Ca2+ fluctuations that occur during membrane damage and repair [5]. Thus, extreme care should be taken in interpreting pro-inflammatory cytokine production in response to CDCs.
Direct cytokine production in response to CDCs is triggered by inflammasome (see Section 2.2), p38, and Ca2+ dependent pathways. Activation of p38 by SLO, PLY, ALO, and VLY leads to IL-8 production [6][7][8]. Activation of p38 also induces secretion of macrophage migration inhibitory factor (MIF) by PLY, which helps reduce the bacterial load [9]. Finally, p38 also stimulates TNFα production after SLO challenge [10]. TNFα is critical for recruiting macrophages during a subcutaneous S. pyogenes infection, which limits bacterial dissemination [11]. CDCs also stimulate IL-6 production. LLO, PLY, and SLY stimulate IL-6, which is Ca2+ dependent for LLO and PLY [12][13][14][15]. Similarly, LLO and PLY trigger IL-1α release and calpain activation in a Ca2+ dependent fashion [3][4]. Overall, CDCs can activate pro-inflammatory cytokine signaling.
The ability of CDCs to directly stimulate TLR signaling pathways remains controversial. While some studies suggest that PLY [16][17], ALO, LLO, SLO, and PFO [18] all trigger TLR4, other studies have not observed TLR4-dependent responses [19][20]. Similarly, NF-ΚB activation, which is downstream of TLRs, is variably reported for CDCs. Some studies observe NF-ΚB activation by CDCs [21][22], while another did not [20]. One potential explanation for the discrepancy is the autocrine and the paracrine effects involving TLRs and/or IL-1 receptor (IL-1R), which also signals through MyD88. LLO-induced NF-ΚB is IL-1R dependent [23]. PLY-induced TNFα production and TLR4 activation were measured at 24 h [24][25], thus TLR4 activation could occur secondary to DAMP release. There are also cell type differences in cytokine production in response to CDCs. For example, PLY induces opposite effects for TNFα production in dendritic cells and macrophages [26]. Overall, TLR activation may be secondary to other effects of CDCs.
2.2. CDCs Activate the Inflammasome
Along with the previously discussed cytokines, CDCs promote IL-1 secretion [25][27][28]. IL-1β is usually secreted following activation of the inflammasome. The inflammasome is a multiprotein complex comprising a cytoplasmic sensory pattern-recognition receptor, the scaffolding protein apoptosis-associated speck-like protein containing a CARD (ASC)/Pycard and an inflammatory caspase (caspase-1 or caspase-11 in mouse, caspases-1,4 or 5 in human) that promotes the inflammatory cell death process pyroptosis and the activation/release of pro-inflammatory cytokines IL-1β and IL-18 (reviewed in [1][29]) (Figure 1). Sensory pattern-recognition receptors that activate the inflammasome include proteins in both the Pyhin-family and the nod-like receptors (NLRs). They sense various pathogen-associated molecular patterns and danger-associated molecular patterns, including membrane perforation by CDCs [1][29]. Membrane perforation is sensed by nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3), presumably indirectly via loss of K+ [30], while cytoplasmic bacterial or mitochondrial DNA is sensed by absent in melanoma 2 (AIM2) [31] (Figure 1). After activation, NLRP3 or AIM2 oligomerize ASC, which recruits and activates caspase-1. Activated caspase-1 then cleaves the pore-forming toxin gasdermin D to promote pyroptosis (Figure 1). Pyroptosis prevents intracellular pathogens from sheltering in the cell and releases pro-inflammatory mediators, including HMGB1, IL-1β, and IL-18 [1][29]. The inflammasome promotes anti-pathogen responses after sensing the CDC challenge.
CDCs activate two inflammasomes. CDCs directly activate the NLRP3 inflammasome via membrane perforation and K+ efflux, and some also indirectly activate AIM2 by facilitating the entry of mitochondrial or bacterial DNA into the cytosol. PFO [32], SLO [33][34], TLO [35], PLY [36][37], LLO [38][39][40], and SLY [41] all activate the NLRP3 inflammasome. LLO-mediated phagosomal rupture and lysosomal permeabilization further activate NLRP3 [39]. Bacteria deficient in LLO [40][42], PLY [36][37], PFO [32], and SLO [34] fail to stimulate IL-1β production. Other toxins, including streptolysin S [43] and C. perfringens α-toxin [32], fail to activate the inflammasome. Thus, CDCs are necessary for pathogen sensing by the NLRP3 inflammasome.
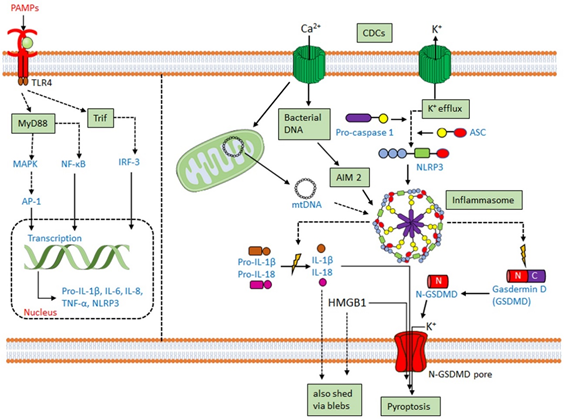
AIM2 activation has functional redundancy with NLRP3 in responding to CDCs. AIM2 can be activated by either bacterial DNA or mitochondrial DNA following L. monocytogenes infection or PLY intoxication [44][45][46][47][48]. AIM2 is also activated by mitochondrial DNA released into the cytosol after cholesterol perturbations [49]. Upregulation of the enzyme cholesterol-25-hydroxylase (Ch25h) by type I or type II interferons (IFNs) produces the regulatory oxysterol, 25-hydroxy-cholesterol [50]. The 25-hydroxy-cholesterol regulates cholesterol biosynthesis, flux, and storage [51][52]. Deletion of Ch25h from macrophages leads to increased IL-1β production in response to LPS-mediated type I interferon (IFN) induction [53]. When IFNs are unable to induce Ch25h, macrophages undergo cholesterol overload and switch to aerobic glycolysis with mitochondrial damage [49]. Mitochondrial damage permits the escape of mitochondrial DNA into the cytosol, activating the AIM2 inflammasome to overproduce IL-1β [49]. Consequently, Ch25h deletion confers AIM2-dependent protection from L. monocytogenes infection on macrophages [49][54].
2.3. CDCs Damage Phagosomes and Permit Phagolysosomal Escape
Phagocytosis is the cellular engulfment process of large particles (>0.5 µm in diameter). It is used by innate immune cells such as macrophages, dendritic cells, and neutrophils to internalize and kill extracellular pathogens [55]. Intracellular pathogens typically hijack phagocytosis to prevent phagosome fusion with the lysosome and may escape into the cytosol. Indeed, L. monocytogenes requires LLO for phagosomal escape and for preventing fusion with lysosomes [56][57][58]. While it is known that transgenic expression of other CDCs such as PFO can promote the escape of L. monocytogenes [59] or Bacillus subtilis [60][61], it is now appreciated that other traditionally extracellular bacteria such as S. pyogenes and C. perfringens rely on CDC-dependent phagosome interference to promote infection [62][63][64][65]. However, phagosomal escape is not perfect [66], thus it is not an all-or-none process. Interestingly, CDCs variably stimulate or impair phagocytosis. LLO stimulates ion flux, which promotes the internalization of L. monocytogenes [67][68]. Conversely, SLO interferes with phagocytosis [69]. However, after membrane damage and repair, compensatory endocytosis is activated to restore homeostasis [70][71]. Before phagosomal escape, LLO, PFO, and PLY may also interfere with acidification in non-macrophages [57]. However, lysosomal membrane permeability stimulates inflammasome activation in macrophages [72][73][74]. Consequently, when PLY interferes with lysosomal acidification in macrophages, it also drives cell death [75]. Interestingly, these events appear to occur independently of phagosome escape. Importantly, in macrophages, LLO permeabilizes phagosomes to small molecules prior to large ones, which led the authors to conclude that LLO forms pores of different sizes, possibly due to insertion of incomplete pores [58]. However, ESCRT-III mediates phagosomal membrane repair [76], thus an alternative explanation is that repair mechanisms limit the extent of damage caused by LLO, preventing the loss of larger molecules. While the structure of LLO is optimized for activity at low pH [77], the host protein gamma-interferon inducible lysosomal thiolreductase (GILT) further activates LLO by reducing the single cysteine in the protein [78]. Thus, CDCs extensively target phagocytosis to evade cell death and promote escape and immune evasion.
2.4. CDC-Mediated Innate Immune Evasion
While CDCs activate several cell defense mechanisms, they also contribute to evading immune activation. Immune evasion is best described for SLO and PLY. CDCs interfere with phagocytosis and inhibit cytokine production. SLO reduces phagocytosis and S. pyogenes killing by neutrophils [69]. Similarly, PLY-stimulated pyroptosis of neutrophils lead to elastase release, which blocks phagocytosis of S. pneumoniae in the inflammasome-defective macrophage cell line Raw264.7 [79]. SLO may stimulate the ubiquitination and the degradation of IL-1β [80]. Similarly, PLY expression reduces maturation of human DCs and pro-IL-1β, IL-8, and IL-12p70 production in response to S. pneumoniae [81]. Finally, PLY, PFO, and SLO can block TNFα production [19][26]. Thus, CDCs promote immune evasion.
2.5. CDCs as an Adaptive Immune Target
While CDCs try to impair macrophages, CDCs are popular targets for antigen presentation by macrophages and other APCs to the adaptive immune system. Bacteriocidal T and B cell responses to LLO are mounted against L. monocytogenes [82][83], LLO-expressing Escherichia coli [84], or B. subtilis [85]. Memory CD4+ T cells respond to PLY [86] and are associated with an absence of S. pneumoniae carriage [87]. Antibodies to ALO [88], PFO [89], or SLY [90] protect mice from lethal infections. Antibodies to CDCs are readily produced [13][82][88][91][92][93]. Anti-SLO and anti-PLY titers can be detected in human serum [93][94], indicating CDCs are robustly antigenic in humans. Importantly, anti-CDC T and B cell responses do not depend on hemolytic activity [92][95], which suggests that non-hemolytic CDC toxoids may serve as useful vaccine targets.
This excerpt is from Thapa et al Toxins (Basel). 2020 Aug 19;12(9):E531. Refer to the article for reference numbers and more information.
References
- Keyel, P.A. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 2014, 69, 136–145.
- Shi, G.; Abbott, K.N.; Wu, W.; Salter, R.D.; Keyel, P.A. Dnase1L3 Regulates Inflammasome-Dependent Cytokine Secretion. Front. Immunol. 2017, 8, 522.
- Fang, R.; Wu, R.; Du, H.; Jin, M.; Liu, Y.; Lei, G.; Jiang, B.; Lei, Z.; Peng, Y.; Nie, K.; et al. Pneumolysin-Dependent Calpain Activation and Interleukin-1alpha Secretion in Macrophages Infected with Streptococcus pneumoniae. Infect. Immun. 2017, 85.
- Dewamitta, S.R.; Nomura, T.; Kawamura, I.; Hara, H.; Tsuchiya, K.; Kurenuma, T.; Shen, Y.; Daim, S.; Yamamoto, T.; Qu, H.; et al. Listeriolysin O-dependent bacterial entry into the cytoplasm is required for calpain activation and interleukin-1 alpha secretion in macrophages infected with Listeria monocytogenes. Infect. Immun. 2010, 78, 1884–1894.
- Heidi Wolfmeier; Roman Schoenauer; Alexander P. Atanassoff; Daniel R. Neill; Aras Kadioglu; Annette Draeger; Eduard B. Babiychuk; Ca2+-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2015, 1853, 2045-2054, 10.1016/j.bbamcr.2014.09.005.
- Shari E. Gelber; Jorge L. Aguilar; Kanako L. T. Lewis; Adam J. Ratner; Functional and Phylogenetic Characterization of Vaginolysin, the Human-Specific Cytolysin from Gardnerella vaginalis. Journal of Bacteriology 2008, 190, 3896-3903, 10.1128/jb.01965-07.
- Adam J. Ratner; Karen R. Hippe; Jorge L. Aguilar; Matthew H. Bender; Aaron L. Nelson; Jeffrey N Weiser; Epithelial Cells Are Sensitive Detectors of Bacterial Pore-forming Toxins. Journal of Biological Chemistry 2006, 281, 12994-12998, 10.1074/jbc.m511431200.
- Jorge L. Aguilar; Ritwij Kulkarni; Tara M. Randis; Sandeep Soman; Alexander Kikuchi; Yuxin Yin; Adam J. Ratner; Phosphatase-Dependent Regulation of Epithelial Mitogen-Activated Protein Kinase Responses to Toxin-Induced Membrane Pores. PLOS ONE 2009, 4, e8076, 10.1371/journal.pone.0008076.
- Rituparna Das; Meredith I. LaRose; Christopher B. Hergott; Lin Leng; Richard Bucala; Jeffrey N. Weiser; Macrophage Migration Inhibitory Factor Promotes Clearance of Pneumococcal Colonization. The Journal of Immunology 2014, 193, 764-772, 10.4049/jimmunol.1400133.
- Stassen, M.; Muller, C.; Richter, C.; Neudorfl, C.; Hultner, L.; Bhakdi, S.; Walev, I.; Schmitt, E. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor alpha by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect. Immun. 2003, 71, 6171–6177.
- Mishalian, I.; Ordan, M.; Peled, A.; Maly, A.; Eichenbaum, M.B.; Ravins, M.; Aychek, T.; Jung, S.; Hanski, E. Recruited macrophages control dissemination of group A Streptococcus from infected soft tissues. J. Immunol. 2011, 187, 6022–6031.
- Tsuchiya, K.; Kawamura, I.; Takahashi, A.; Nomura, T.; Kohda, C.; Mitsuyama, M. Listeriolysin O-induced membrane permeation mediates persistent interleukin-6 production in Caco-2 cells during Listeria monocytogenes infection in vitro. Infect. Immun. 2005, 73, 3869–3877. [Google Scholar] [CrossRef]
- Lun, S.; Perez-Casal, J.; Connor, W.; Willson, P.J. Role of suilysin in pathogenesis of Streptococcus suis capsular serotype 2. Microb. Pathog. 2003, 34, 27–37.
- Rijneveld, A.W.; van den Dobbelsteen, G.P.; Florquin, S.; Standiford, T.J.; Speelman, P.; van Alphen, L.; van der Poll, T. Roles of interleukin-6 and macrophage inflammatory protein-2 in pneumolysin-induced lung inflammation in mice. J. Infect. Dis. 2002, 185, 123–126.
- Rose, F.; Zeller, S.A.; Chakraborty, T.; Domann, E.; Machleidt, T.; Kronke, M.; Seeger, W.; Grimminger, F.; Sibelius, U. Human endothelial cell activation and mediator release in response to Listeria monocytogenes virulence factors. Infect. Immun. 2001, 69, 897–905.
- Malley, R.; Henneke, P.; Morse, S.C.; Cieslewicz, M.J.; Lipsitch, M.; Thompson, C.M.; Kurt-Jones, E.; Paton, J.C.; Wessels, M.R.; Golenbock, D.T. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA 2003, 100, 1966–1971.
- Dessing, M.C.; Hirst, R.A.; de Vos, A.F.; van der Poll, T. Role of Toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS ONE 2009, 4, e7993.
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655.
- Bhattacharjee, P.; Keyel, P.A. Cholesterol-dependent cytolysins impair pro-inflammatory macrophage responses. Sci. Rep. 2018, 8.
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Nunez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238.
- Fickl, H.; Cockeran, R.; Steel, H.C.; Feldman, C.; Cowan, G.; Mitchell, T.J.; Anderson, R. Pneumolysin-mediated activation of NFkappaB in human neutrophils is antagonized by docosahexaenoic acid. Clin. Exp. Immunol. 2005, 140, 274–281.
- Kayal, S.; Lilienbaum, A.; Poyart, C.; Memet, S.; Israel, A.; Berche, P. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: Activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 1999, 31, 1709–1722.
- Kayal, S.; Lilienbaum, A.; Join-Lambert, O.; Li, X.; Israel, A.; Berche, P. Listeriolysin O secreted by Listeria monocytogenes induces NF-kappaB signalling by activating the IkappaB kinase complex. Mol. Microbiol. 2002, 44, 1407–1419.
- Shoma, S.; Tsuchiya, K.; Kawamura, I.; Nomura, T.; Hara, H.; Uchiyama, R.; Daim, S.; Mitsuyama, M. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae In Vitro: A novel function of pneumolysin in caspase-1 activation. Infect. Immun. 2008, 76, 1547–1557.
- Houldsworth, S.; Andrew, P.W.; Mitchell, T.J. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect. Immun. 1994, 62, 1501–1503.
- Subramanian, K.; Neill, D.R.; Malak, H.A.; Spelmink, L.; Khandaker, S.; Dalla Libera Marchiori, G.; Dearing, E.; Kirby, A.; Yang, M.; Achour, A.; et al. Pneumolysin binds to the mannose receptor C type 1 (MRC-1) leading to anti-inflammatory responses and enhanced pneumococcal survival. Nat. Microbiol. 2019, 4, 62–70.
- Nishibori, T.; Xiong, H.; Kawamura, I.; Arakawa, M.; Mitsuyama, M. Induction of cytokine gene expression by listeriolysin O and roles of macrophages and NK cells. Infect. Immun. 1996, 64, 3188–3195.
- Hackett, S.P.; Stevens, D.L. Streptococcal toxic shock syndrome: Synthesis of tumor necrosis factor and interleukin-1 by monocytes stimulated with pyrogenic exotoxin A and streptolysin O. J. Infect. Dis. 1992, 165, 879–885.
- Petr Broz; Vishva M. Dixit; Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews Immunology 2016, 16, 407-420, 10.1038/nri.2016.58.
- Raúl Muñoz-Planillo; Peter Kuffa; Giovanny Martínez-Colón; Brenna L. Smith; Thekkelnaycke M. Rajendiran; Gabriel Núñez; K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142-1153, 10.1016/j.immuni.2013.05.016.
- Veit Hornung; Andrea Ablasser; Marie Charrel-Dennis; Franz G. Bauernfeind; Gabor Horvath; Daniel. R. Caffrey; Eicke Latz; Katherine A. Fitzgerald; AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514-518, 10.1038/nature07725.
- Kiyonobu Yamamura; Hiroshi Ashida; Tokuju Okano; Ryo Kinoshita-Daitoku; Shiho Suzuki; Kaori Ohtani; Miwako Hamagaki; Tohru Ikeda; Toshihiko Suzuki; Inflammasome Activation Induced by Perfringolysin O of Clostridium perfringens and Its Involvement in the Progression of Gas Gangrene. Frontiers in Microbiology 2019, 10, 2406, 10.3389/fmicb.2019.02406.
- Peter A. Keyel; Robyn Roth; Wayne M. Yokoyama; John E. Heuser; Russell D Salter; Reduction of Streptolysin O (SLO) Pore-Forming Activity Enhances Inflammasome Activation. Toxins 2013, 5, 1105-1118, 10.3390/toxins5061105.
- Jürgen Harder; Luigi Franchi; Raúl Muñoz-Planillo; Jong-Hwan Park; Thornik Reimer; Gabriel Núñez; Activation of the Nlrp3 Inflammasome byStreptococcus pyogenesRequires Streptolysin O and NF-κB Activation but Proceeds Independently of TLR Signaling and P2X7 Receptor. The Journal of Immunology 2009, 183, 5823-5829, 10.4049/jimmunol.0900444.
- Jessica Chu; L. Michael Thomas; Simon C. Watkins; Luigi Franchi; Gabriel Núñez; Russell D. Salter; Cholesterol-dependent cytolysins induce rapid release of mature IL-1β from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. Journal of Leukocyte Biology 2009, 86, 1227-1238, 10.1189/jlb.0309164.
- Martin Witzenrath; Florence Pache; Daniel Lorenz; Uwe Koppe; Birgitt Gutbier; Christoph Tabeling; Katrin Reppe; Karolin Meixenberger; Anca Dorhoi; Jiangtao T. Ma; et al.Ashleigh HolmesGeorge TrendelenburgMarkus M. HeimesaatStefan BereswillMark Van Der LindenJürg TschoppTimothy J. MitchellNorbert SuttorpBastian Opitz The NLRP3 Inflammasome Is Differentially Activated by Pneumolysin Variants and Contributes to Host Defense in Pneumococcal Pneumonia. The Journal of Immunology 2011, 187, 434-440, 10.4049/jimmunol.1003143.
- Edel A. McNeela; Áine Burke; Daniel R. Neill; Cathy Baxter; Vitor E. Fernandes; Daniela Ferreira; Sarah Smeaton; Rana El-Rachkidy; Rachel M. McLoughlin; Andres Mori; et al.Barry MoranKatherine A. FitzgeraldJürg TschoppVirginie PetrilliPeter W. AndrewAras KadiogluEd C. Lavelle Pneumolysin Activates the NLRP3 Inflammasome and Promotes Proinflammatory Cytokines Independently of TLR4. PLOS Pathogens 2010, 6, e1001191, 10.1371/journal.ppat.1001191.
- Warren, S.E.; Mao, D.P.; Rodriguez, A.E.; Miao, E.A.; Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 2008, 180, 7558–7564.
- Meixenberger, K.; Pache, F.; Eitel, J.; Schmeck, B.; Hippenstiel, S.; Slevogt, H.; N’Guessan, P.; Witzenrath, M.; Netea, M.G.; Chakraborty, T.; et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 2010, 184, 922–930.
- Hara, H.; Tsuchiya, K.; Nomura, T.; Kawamura, I.; Shoma, S.; Mitsuyama, M. Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J. Immunol. 2008, 180, 7859–7868.
- Lavagna, A.; Auger, J.P.; Dumesnil, A.; Roy, D.; Girardin, S.E.; Gisch, N.; Segura, M.; Gottschalk, M. Interleukin-1 signaling induced by Streptococcus suis serotype 2 is strain-dependent and contributes to bacterial clearance and inflammation during systemic disease in a mouse model of infection. Vet. Res. 2019, 50, 52.
- Nesrin Özören; Junya Masumoto; Luigi Franchi; Thirumala-Devi Kanneganti; Mathilde Body-Malapel; Ilkim Ertürk; Rajesh Jagirdar; Li Zhu; Naohiro Inohara; John Bertin; et al.Anthony CoyleEthan P. GrantGabriel Núñez Distinct Roles of TLR2 and the Adaptor ASC in IL-1β/IL-18 Secretion in Response toListeria monocytogenes. The Journal of Immunology 2006, 176, 4337-4342, 10.4049/jimmunol.176.7.4337.
- Anjuli M. Timmer; John C. Timmer; Morgan A. Pence; Li-Chung Hsu; Mariam Ghochani; Terrence G. Frey; Michael Karin; Guy S. Salvesen; Victor Nizet; Streptolysin O Promotes Group AStreptococcusImmune Evasion by Accelerated Macrophage Apoptosis. Journal of Biological Chemistry 2008, 284, 862-871, 10.1074/jbc.m804632200.
- Johann Braun; Olaf Hoffmann; Miriam Schickhaus; Dorette Freyer; Emilie Dagand; Daniela Bermpohl; Tim J. Mitchell; Ingo Bechmann; Joerg R. Weber; Pneumolysin Causes Neuronal Cell Death through Mitochondrial Damage. Infection and Immunity 2007, 75, 4245-4254, 10.1128/iai.00031-07.
- Jamie K. Lemon; Jeffrey N Weiser; Jeffrey R. Kugelman; Mariano Sanchez-Lockhart; Kristian G. Andersen; Stephen Gire; Daniel J. Park; Rachel Sealfon; Aaron E. Lin; Shirlee Wohl; et al.Pardis C. SabetiJens H. KuhnGustavo F. Palacios Degradation Products of the Extracellular Pathogen Streptococcus pneumoniae Access the Cytosol via Its Pore-Forming Toxin. mBio 2015, 6, e02227-14, 10.1128/mbio.02110-14.
- Fang, R.; Tsuchiya, K.; Kawamura, I.; Shen, Y.; Hara, H.; Sakai, S.; Yamamoto, T.; Fernandes-Alnemri, T.; Yang, R.; Hernandez-Cuellar, E.; et al. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 2011, 187, 4890–4899.
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402.
- Kim, S.; Bauernfeind, F.; Ablasser, A.; Hartmann, G.; Fitzgerald, K.A.; Latz, E.; Hornung, V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 2010, 40, 1545–1551.
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e1011.
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087.
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518.
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157.
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684.
- Abrams, M.E.; Johnson, K.A.; Perelman, S.S.; Zhang, L.S.; Endapally, S.; Mar, K.B.; Thompson, B.M.; McDonald, J.G.; Schoggins, J.W.; Radhakrishnan, A.; et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 2020, 5, 929–942.
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98.
- Portnoy, D.A.; Jacks, P.S.; Hinrichs, D.J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1988, 167, 1459–1471.
- Malet, J.K.; Cossart, P.; Ribet, D. Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol-dependent cytolysins. Cell. Microbiol. 2017, 19, e12682.
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell. Microbiol. 2006, 8, 781–792.
- Jones, S.; Portnoy, D.A. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 1994, 62, 5608–5613.
- Portnoy, D.A.; Tweten, R.K.; Kehoe, M.; Bielecki, J. Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect. Immun. 1992, 60, 2710–2717.
- Bielecki, J.; Youngman, P.; Connelly, P.; Portnoy, D.A. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 1990, 345, 175–176.
- O’Brien, D.K.; Melville, S.B. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect. Immun. 2004, 72, 5204–5215.
- Bastiat-Sempe, B.; Love, J.F.; Lomayesva, N.; Wessels, M.R. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio 2014, 5, e01690-14.
- Hickey, M.J.; Kwan, R.Y.; Awad, M.M.; Kennedy, C.L.; Young, L.F.; Hall, P.; Cordner, L.M.; Lyras, D.; Emmins, J.J.; Rood, J.I. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008, 4, e1000045.
- O’Neill, A.M.; Thurston, T.L.; Holden, D.W. Cytosolic Replication of Group A Streptococcus in Human Macrophages. mBio 2016, 7, e00020-16.
- De Chastellier, C.; Berche, P. Fate of Listeria monocytogenes in murine macrophages: Evidence for simultaneous killing and survival of intracellular bacteria. Infect. Immun. 1994, 62, 543–553.
- Vadia, S.; Arnett, E.; Haghighat, A.C.; Wilson-Kubalek, E.M.; Tweten, R.K.; Seveau, S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 2011, 7, e1002356.
- Dramsi, S.; Cossart, P. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect. Immun. 2003, 71, 3614–3618.
- Gabriele Sierig; Colette Cywes; Michael R Wessels; Cameron D Ashbaugh; Cytotoxic effects of streptolysin o and streptolysin s enhance the virulence of poorly encapsulated group a streptococci.. Infection and Immunity 2003, 71, 446-455.
- Matthew Romero; Michelle Keyel; Guilan Shi; Pushpak Bhattacharjee; Robyn Roth; John E Heuser; Peter A Keyel; Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death & Differentiation 2017, 24, 798-808, 10.1038/cdd.2017.11.
- Matthias Corrotte; Patricia E Almeida; Christina Tam; Thiago Castro-Gomes; Maria Cecilia Fernandes; Bryan A Millis; Mauro Cortez; Heather Miller; Wenxia Song; Timothy K Maugel; et al.Norma W. Andrews Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926, 10.7554/elife.00926.
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238.
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856.
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865.
- Martin A. Bewley; Michael Naughton; Julie Preston; Andrea Mitchell; Ashleigh Holmes; Helen M. Marriott; Robert C. Read; Timothy J. Mitchell; Moira K. B. Whyte; David H. Dockrell; et al. Pneumolysin Activates Macrophage Lysosomal Membrane Permeabilization and Executes Apoptosis by Distinct Mechanisms without Membrane Pore Formation. mBio 2014, 5, e01710-14-14, 10.1128/mbio.01710-14.
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360.
- Schuerch, D.W.; Wilson-Kubalek, E.M.; Tweten, R.K. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. USA 2005, 102, 12537–12542.
- Singh, R.; Jamieson, A.; Cresswell, P. GILT is a critical host factor for Listeria monocytogenes infection. Nature 2008, 455, 1244–1247.
- Hisanori Domon; Masataka Oda; Tomoki Maekawa; Kosuke Nagai; Wataru Takeda; Yutaka Terao; Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Scientific Reports 2016, 6, 38013, 10.1038/srep38013.
- Hancz, D.; Westerlund, E.; Valfridsson, C.; Aemero, G.M.; Bastiat-Sempe, B.; Orning, P.; Lien, E.; Wessels, M.R.; Persson, J.J. Streptolysin O Induces the Ubiquitination and Degradation of Pro-IL-1beta. J. Innate Immun. 2019, 11, 457–468.
- Littmann, M.; Albiger, B.; Frentzen, A.; Normark, S.; Henriques-Normark, B.; Plant, L. Streptococcus pneumoniae evades human dendritic cell surveillance by pneumolysin expression. EMBO Mol. Med. 2009, 1, 211–222.
- Edelson, B.T.; Cossart, P.; Unanue, E.R. Cutting edge: Paradigm revisited: Antibody provides resistance to Listeria infection. J. Immunol. 1999, 163, 4087–4090.
- Pamer, E.G.; Harty, J.T.; Bevan, M.J. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature 1991, 353, 852–855.
- Hu, P.Q.; Tuma-Warrino, R.J.; Bryan, M.A.; Mitchell, K.G.; Higgins, D.E.; Watkins, S.C.; Salter, R.D. Escherichia coli expressing recombinant antigen and listeriolysin O stimulate class I-restricted CD8+ T cells following uptake by human APC. J. Immunol. 2004, 172, 1595–1601.
- Bouwer, H.G.; Nelson, C.S.; Gibbins, B.L.; Portnoy, D.A.; Hinrichs, D.J. Listeriolysin O is a target of the immune response to Listeria monocytogenes. J. Exp. Med. 1992, 175, 1467–1471.
- Mureithi, M.W.; Finn, A.; Ota, M.O.; Zhang, Q.; Davenport, V.; Mitchell, T.J.; Williams, N.A.; Adegbola, R.A.; Heyderman, R.S. T cell memory response to pneumococcal protein antigens in an area of high pneumococcal carriage and disease. J. Infect. Dis. 2009, 200, 783–793.
- Zhang, Q.; Bagrade, L.; Bernatoniene, J.; Clarke, E.; Paton, J.C.; Mitchell, T.J.; Nunez, D.A.; Finn, A. Low CD4 T cell immunity to pneumolysin is associated with nasopharyngeal carriage of pneumococci in children. J. Infect. Dis. 2007, 195, 1194–1202.
- Nakouzi, A.; Rivera, J.; Rest, R.F.; Casadevall, A. Passive administration of monoclonal antibodies to anthrolysin O prolong survival in mice lethally infected with Bacillus anthracis. BMC Microbiol. 2008, 8, 159.
- Stevens, D.L.; Tweten, R.K.; Awad, M.M.; Rood, J.I.; Bryant, A.E. Clostridial gas gangrene: Evidence that alpha and theta toxins differentially modulate the immune response and induce acute tissue necrosis. J. Infect. Dis. 1997, 176, 189–195.
- A A Jacobs; P L Loeffen; A J Van Den Berg; P K Storm; Identification, purification, and characterization of a thiol-activated hemolysin (suilysin) of Streptococcus suis.. Infection and Immunity 1994, 62, 1742-1748, 10.1128/iai.62.5.1742-1748.1994.
- Chiarot, E.; Faralla, C.; Chiappini, N.; Tuscano, G.; Falugi, F.; Gambellini, G.; Taddei, A.; Capo, S.; Cartocci, E.; Veggi, D.; et al. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio 2013, 4, e00387-12.
- Jacobs, A.A.; van den Berg, A.J.; Loeffen, P.L. Protection of experimentally infected pigs by suilysin, the thiol-activated haemolysin of Streptococcus suis. Vet. Rec. 1996, 139, 225–228.
- Musher, D.M.; Phan, H.M.; Baughn, R.E. Protection against bacteremic pneumococcal infection by antibody to pneumolysin. J. Infect. Dis. 2001, 183, 827–830.
- Wade, K.R.; Hotze, E.M.; Briles, D.E.; Tweten, R.K. Mouse, but not human, ApoB-100 lipoprotein cholesterol is a potent innate inhibitor of Streptococcus pneumoniae pneumolysin. PLoS Pathog. 2014, 10, e1004353.
- Carrero, J.A.; Vivanco-Cid, H.; Unanue, E.R. Listeriolysin o is strongly immunogenic independently of its cytotoxic activity. PLoS ONE 2012, 7, e32310.



